当前位置:
X-MOL 学术
›
Nat. Microbiol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth's biomes.
Nature Microbiology ( IF 20.5 ) Pub Date : 2019-07-22 , DOI: 10.1038/s41564-019-0510-x Simon Roux 1 , Mart Krupovic 2 , Rebecca A Daly 3 , Adair L Borges 4 , Stephen Nayfach 1 , Frederik Schulz 1 , Allison Sharrar 5 , Paula B Matheus Carnevali 5 , Jan-Fang Cheng 1 , Natalia N Ivanova 1 , Joseph Bondy-Denomy 4, 6 , Kelly C Wrighton 3 , Tanja Woyke 1 , Axel Visel 1 , Nikos C Kyrpides 1 , Emiley A Eloe-Fadrosh 1
Nature Microbiology ( IF 20.5 ) Pub Date : 2019-07-22 , DOI: 10.1038/s41564-019-0510-x Simon Roux 1 , Mart Krupovic 2 , Rebecca A Daly 3 , Adair L Borges 4 , Stephen Nayfach 1 , Frederik Schulz 1 , Allison Sharrar 5 , Paula B Matheus Carnevali 5 , Jan-Fang Cheng 1 , Natalia N Ivanova 1 , Joseph Bondy-Denomy 4, 6 , Kelly C Wrighton 3 , Tanja Woyke 1 , Axel Visel 1 , Nikos C Kyrpides 1 , Emiley A Eloe-Fadrosh 1
Affiliation
|
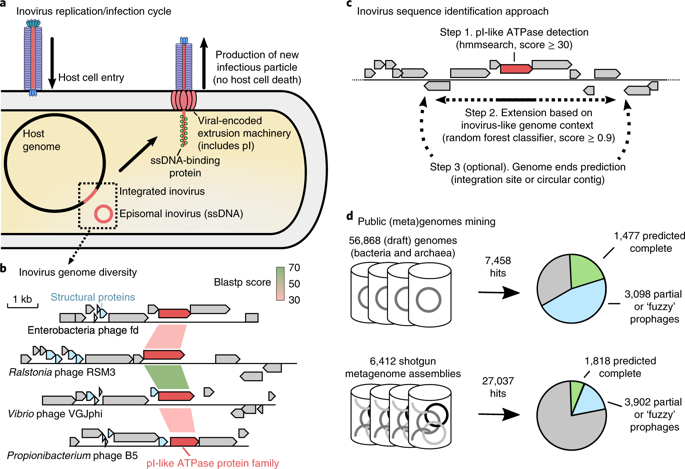
Bacteriophages from the Inoviridae family (inoviruses) are characterized by their unique morphology, genome content and infection cycle. One of the most striking features of inoviruses is their ability to establish a chronic infection whereby the viral genome resides within the cell in either an exclusively episomal state or integrated into the host chromosome and virions are continuously released without killing the host. To date, a relatively small number of inovirus isolates have been extensively studied, either for biotechnological applications, such as phage display, or because of their effect on the toxicity of known bacterial pathogens including Vibrio cholerae and Neisseria meningitidis. Here, we show that the current 56 members of the Inoviridae family represent a minute fraction of a highly diverse group of inoviruses. Using a machine learning approach leveraging a combination of marker gene and genome features, we identified 10,295 inovirus-like sequences from microbial genomes and metagenomes. Collectively, our results call for reclassification of the current Inoviridae family into a viral order including six distinct proposed families associated with nearly all bacterial phyla across virtually every ecosystem. Putative inoviruses were also detected in several archaeal genomes, suggesting that, collectively, members of this supergroup infect hosts across the domains Bacteria and Archaea. Finally, we identified an expansive diversity of inovirus-encoded toxin-antitoxin and gene expression modulation systems, alongside evidence of both synergistic (CRISPR evasion) and antagonistic (superinfection exclusion) interactions with co-infecting viruses, which we experimentally validated in a Pseudomonas model. Capturing this previously obscured component of the global virosphere may spark new avenues for microbial manipulation approaches and innovative biotechnological applications.
中文翻译:

隐性猪病毒在整个地球生物群系的细菌和古细菌中普遍存在。
来自Inoviridae家族的噬菌体(inoviruses)的特征在于其独特的形态,基因组含量和感染周期。禽病毒最显着的特征之一是它们能够建立慢性感染的能力,从而病毒基因组以完全游离状态或整合到宿主染色体中的方式位于细胞内,并且病毒颗粒不断释放而不会杀死宿主。迄今为止,已经针对生物技术应用,例如噬菌体展示,或由于它们对包括霍乱弧菌和脑膜炎奈瑟氏球菌在内的已知细菌病原体的毒性影响,已经对相对少量的猪病毒分离物进行了广泛的研究。在这里,我们显示了Inoviridae家族的当前56个成员代表了高度多样化的inoviruses组的一小部分。使用结合标记物基因和基因组特征的机器学习方法,我们从微生物基因组和元基因组中鉴定了10,295种禽病毒样序列。总的来说,我们的结果要求将当前的Inoviridae家族重新分类为病毒顺序,包括与几乎每个生态系统中几乎所有细菌门相关的六个不同的拟议家族。在几个古细菌基因组中也检测到推定的禽病毒,这表明该超群成员共同感染了细菌和古细菌领域的宿主。最后,我们确定了感染病毒编码的毒素-抗毒素和基因表达调节系统的广泛多样性,以及与共感染病毒协同(CRISPR逃避)和拮抗(排除超级感染)相互作用的证据,我们在假单胞菌模型中通过实验验证了这一点。捕获以前被遮盖的全球病毒圈的组件可能会为微生物操纵方法和创新的生物技术应用程序开辟新的途径。
更新日期:2019-07-22
中文翻译:
隐性猪病毒在整个地球生物群系的细菌和古细菌中普遍存在。
来自Inoviridae家族的噬菌体(inoviruses)的特征在于其独特的形态,基因组含量和感染周期。禽病毒最显着的特征之一是它们能够建立慢性感染的能力,从而病毒基因组以完全游离状态或整合到宿主染色体中的方式位于细胞内,并且病毒颗粒不断释放而不会杀死宿主。迄今为止,已经针对生物技术应用,例如噬菌体展示,或由于它们对包括霍乱弧菌和脑膜炎奈瑟氏球菌在内的已知细菌病原体的毒性影响,已经对相对少量的猪病毒分离物进行了广泛的研究。在这里,我们显示了Inoviridae家族的当前56个成员代表了高度多样化的inoviruses组的一小部分。使用结合标记物基因和基因组特征的机器学习方法,我们从微生物基因组和元基因组中鉴定了10,295种禽病毒样序列。总的来说,我们的结果要求将当前的Inoviridae家族重新分类为病毒顺序,包括与几乎每个生态系统中几乎所有细菌门相关的六个不同的拟议家族。在几个古细菌基因组中也检测到推定的禽病毒,这表明该超群成员共同感染了细菌和古细菌领域的宿主。最后,我们确定了感染病毒编码的毒素-抗毒素和基因表达调节系统的广泛多样性,以及与共感染病毒协同(CRISPR逃避)和拮抗(排除超级感染)相互作用的证据,我们在假单胞菌模型中通过实验验证了这一点。捕获以前被遮盖的全球病毒圈的组件可能会为微生物操纵方法和创新的生物技术应用程序开辟新的途径。






























 京公网安备 11010802027423号
京公网安备 11010802027423号