当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Interplay of Ring Puckering and Hydrogen Bonding in Deoxyribonucleosides
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-07-19 00:00:00 , DOI: 10.1021/acs.jpca.9b05452 Siying Lyu 1 , Nassim Beiranvand 1 , Marek Freindorf 1 , Elfi Kraka 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-07-19 00:00:00 , DOI: 10.1021/acs.jpca.9b05452 Siying Lyu 1 , Nassim Beiranvand 1 , Marek Freindorf 1 , Elfi Kraka 1
Affiliation
|
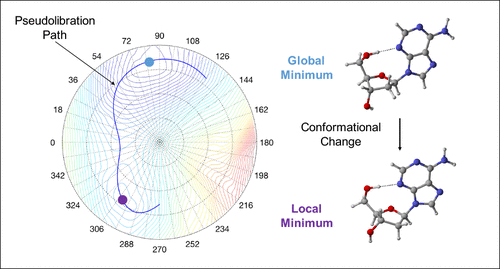
The Cremer–Pople ring puckering analysis and the Konkoli–Cremer local mode analysis supported by the topological analysis of the electron density were applied for the first comprehensive analysis of the interplay between deoxyribose ring puckering and intramolecular H-bonding in 2′-deoxycytidine, 2′-deoxyadenosine, 2′-deoxythymidine, and 2′-deoxyguanosine. We mapped for each deoxyribonucleoside the complete conformational energy surface and the corresponding pseudorotation path. We found only incomplete pseudorotation cycles, caused by ring inversion, which we coined as pseudolibration paths. On each pseudolibration path a global and a local minimum separated by a transition state were identified. The investigation of H-bond free deoxyribonucleoside analogs revealed that removal of the H-bond does not restore the full conformational flexibility of the sugar ring. Our work showed that ring puckering predominantly determines the conformational energy; the larger the puckering amplitude, the lower the conformational energy. In contrast no direct correlation between conformational energy and H-bond strength was found. The longest and weakest H-bonds are located in the local minimum region, whereas the shortest and strongest H-bonds are located outside the global and local minimum regions at the turning points of the pseudolibration paths, i.e., H-bonding determines the shape and length of the pseudolibration paths. In addition to the H-bond strength, we evaluated the covalent/electrostatic character of the H-bonds applying the Cremer–Kraka criterion of covalent bonding. H-bonding in the puric bases has a more covalent character whereas in the pyrimidic bases the H-bond character is more electrostatic. We investigated how the mutual orientation of the CH2OH group and the base influences H-bond formation via two geometrical parameters describing the rotation of the substituents perpendicular to the sugar ring and their tilting relative to the ring center. According to our results, rotation is more important for H-bond formation. In addition we assessed the influence of the H-bond acceptor, the lone pair (N, respectively O), via the delocalization energy. We found larger delocalization energies corresponding to stronger H-bonds for the puric bases. The global minimum conformation of 2′-deoxyguanosine has the strongest H-bond of all conformers investigated in this work with a bond strength of 0.436 which is even stronger than the H-bond in the water dimer (0.360). The application of our new analysis to DNA deoxyribonucleotides and to unnatural base pairs, which have recently drawn a lot of attention, is in progress.
中文翻译:

脱氧核糖核苷的环起皱和氢键相互作用
电子密度拓扑分析支持的Cremer-Pople环褶皱分析和Konkoli-Cremer局域模分析被用于2'-脱氧胞苷2中脱氧核糖环褶皱与分子内H键相互作用的首次综合分析。 '-脱氧腺苷,2'-脱氧胸苷和2'-脱氧鸟苷。我们为每个脱氧核糖核苷绘制了完整的构象能表面和相应的假旋转路径。我们仅发现由环反转引起的不完整的伪旋转循环,我们将其称为伪解放路径。在每个伪解放路径上,均确定了由过渡状态分隔的全局最小值和局部最小值。对无H键的脱氧核糖核苷类似物的研究表明,除去H键不能恢复糖环的完整构象柔性。我们的工作表明,起皱主要决定构象能量;反之亦然。褶皱幅度越大,构象能量越低。相反,未发现构象能与氢键强度之间存在直接关系。最长和最弱的H键位于局部最小区域,而最短和最强的H键位于伪解放路径的转折点处的全局和局部最小区域之外,即,H键决定形状和形状。伪解放路径的长度。除了氢键强度 我们使用Cromer-Kraka共价键准则评估了H键的共价/静电特性。嘌呤碱基中的H键具有更强的共价性,而在嘧啶碱基中,H键具有更强的静电性。我们调查了CH的相互定位2个OH基团和碱通过两个几何参数影响H键的形成,所述两个几何参数描述了垂直于糖环的取代基的旋转及其相对于环中心的倾斜。根据我们的结果,旋转对于氢键的形成更为重要。另外,我们通过离域能量评估了H键受体,孤对(N和O)的影响。我们发现较大的离域能对应于纯净碱基的更强氢键。2'-脱氧鸟苷的整体最小构象在这项研究的所有构象异构体中具有最强的氢键,其结合强度为0.436,甚至比水二聚体中的氢键(0.360)更强。我们的新分析方法在DNA脱氧核糖核苷酸和非天然碱基对中的应用,最近引起了很多关注,
更新日期:2019-07-19
中文翻译:
脱氧核糖核苷的环起皱和氢键相互作用
电子密度拓扑分析支持的Cremer-Pople环褶皱分析和Konkoli-Cremer局域模分析被用于2'-脱氧胞苷2中脱氧核糖环褶皱与分子内H键相互作用的首次综合分析。 '-脱氧腺苷,2'-脱氧胸苷和2'-脱氧鸟苷。我们为每个脱氧核糖核苷绘制了完整的构象能表面和相应的假旋转路径。我们仅发现由环反转引起的不完整的伪旋转循环,我们将其称为伪解放路径。在每个伪解放路径上,均确定了由过渡状态分隔的全局最小值和局部最小值。对无H键的脱氧核糖核苷类似物的研究表明,除去H键不能恢复糖环的完整构象柔性。我们的工作表明,起皱主要决定构象能量;反之亦然。褶皱幅度越大,构象能量越低。相反,未发现构象能与氢键强度之间存在直接关系。最长和最弱的H键位于局部最小区域,而最短和最强的H键位于伪解放路径的转折点处的全局和局部最小区域之外,即,H键决定形状和形状。伪解放路径的长度。除了氢键强度 我们使用Cromer-Kraka共价键准则评估了H键的共价/静电特性。嘌呤碱基中的H键具有更强的共价性,而在嘧啶碱基中,H键具有更强的静电性。我们调查了CH的相互定位2个OH基团和碱通过两个几何参数影响H键的形成,所述两个几何参数描述了垂直于糖环的取代基的旋转及其相对于环中心的倾斜。根据我们的结果,旋转对于氢键的形成更为重要。另外,我们通过离域能量评估了H键受体,孤对(N和O)的影响。我们发现较大的离域能对应于纯净碱基的更强氢键。2'-脱氧鸟苷的整体最小构象在这项研究的所有构象异构体中具有最强的氢键,其结合强度为0.436,甚至比水二聚体中的氢键(0.360)更强。我们的新分析方法在DNA脱氧核糖核苷酸和非天然碱基对中的应用,最近引起了很多关注,






























 京公网安备 11010802027423号
京公网安备 11010802027423号