当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Tungsten–Ligand Bond Strengths for 2p Elements Including σ- and π-Bond Strength Components, A Density Functional Theory and ab Initio Study
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-06-03 00:00:00 , DOI: 10.1021/acs.jpca.9b03272 Catherine A. Moulder 1 , Kristina Kafle 1 , Thomas R. Cundari 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-06-03 00:00:00 , DOI: 10.1021/acs.jpca.9b03272 Catherine A. Moulder 1 , Kristina Kafle 1 , Thomas R. Cundari 1
Affiliation
|
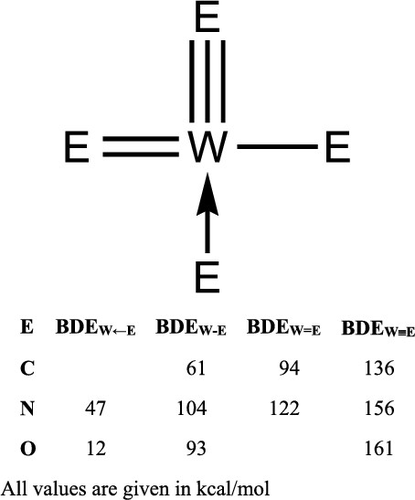
Three WVI crystal structures with multifarious metal–ligand bond types are used to theoretically predict homolytic metal–element bond enthalpies with 11 popular DFT functionals, MP2 wave function methods, and four common valence basis set/pseudopotentials in order to evaluate the accuracy and precision of the resultant bond enthalpy data. To our knowledge, for the first time, estimates of component metal–ligand σ- and π-bond strengths are computed. The WE (E = C, N, O) bond enthalpies have the consistent trend σ > second π > first π. In contrast, the element–element BDE trend for the 2p homologues is second π > first π > σ for nitrogen and oxygen, and σ > first π > second π for carbon. These differences may underpin the differences in stability trends and thus reactivity behavior for metal–element multiple bonds as compared to the element–element multiple bonds, and metal–element triple bonds versus their corresponding double bonded counterparts. For example, Odom et al. show that MeI nucleophilically attacks at the imide (M═N) rather than the nitride (M ≡ N) ligand; the relative π-bond strengths derived herein provide a thermodynamic rationalization for this site preference. In this study, it is deduced from the calculated thermodynamics that the W–oxo ligand is more congruous with a triple bond than a double bond, consistent with the bonding model set forth in the seminal 1961 Ballhausen–Gray paper.
中文翻译:

2p元素的钨-配体键合强度,包括σ和π键强度分量,密度泛函理论和从头算研究
三W VI具有多种金属-配体键类型的晶体结构可用于理论上预测具有11种流行的DFT官能团,MP2波函数方法和四种常用化合价基组/拟电位的均溶金属-元素键焓,以便评估所得结果的准确性和精度键焓数据。据我们所知,这是首次计算出成分金属配体的σ和π键强度的估算值。WE(E = C,N,O)键焓具有一致的趋势σ>第二π>第一π。相反,对于2p同源物,元素-元素的BDE趋势是氮和氧的第二π>第一π>σ,碳的σ>第一π>第二π。这些差异可能是由于金属元素多重键与元素元素多重键以及金属元素三键与对应的双键对应物相比在稳定性趋势和反应行为上的差异所致。例如,奥多姆等。表明MeI亲核性地攻击酰亚胺(M═N)而不是氮化物(M≡N)配体。本文推导的相对π键强度为该位点偏好提供了热力学合理性。在这项研究中,从计算的热力学推论得出,W-氧代配体与三键的结合比与双键的结合更紧密,这与1961年Ballhausen-Gray论文中提出的键合模型一致。
更新日期:2019-06-03
中文翻译:
2p元素的钨-配体键合强度,包括σ和π键强度分量,密度泛函理论和从头算研究
三W VI具有多种金属-配体键类型的晶体结构可用于理论上预测具有11种流行的DFT官能团,MP2波函数方法和四种常用化合价基组/拟电位的均溶金属-元素键焓,以便评估所得结果的准确性和精度键焓数据。据我们所知,这是首次计算出成分金属配体的σ和π键强度的估算值。WE(E = C,N,O)键焓具有一致的趋势σ>第二π>第一π。相反,对于2p同源物,元素-元素的BDE趋势是氮和氧的第二π>第一π>σ,碳的σ>第一π>第二π。这些差异可能是由于金属元素多重键与元素元素多重键以及金属元素三键与对应的双键对应物相比在稳定性趋势和反应行为上的差异所致。例如,奥多姆等。表明MeI亲核性地攻击酰亚胺(M═N)而不是氮化物(M≡N)配体。本文推导的相对π键强度为该位点偏好提供了热力学合理性。在这项研究中,从计算的热力学推论得出,W-氧代配体与三键的结合比与双键的结合更紧密,这与1961年Ballhausen-Gray论文中提出的键合模型一致。






























 京公网安备 11010802027423号
京公网安备 11010802027423号