当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Chemical Dynamics Simulations on Association and Ensuing Dissociation of a Benzene–Hexafluorobenzene Molecular System
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-05-30 00:00:00 , DOI: 10.1021/acs.jpca.9b02332 Himashree Mahanta 1 , Daradi Baishya 1 , Sk. Samir Ahamed 1 , Amit K. Paul 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-05-30 00:00:00 , DOI: 10.1021/acs.jpca.9b02332 Himashree Mahanta 1 , Daradi Baishya 1 , Sk. Samir Ahamed 1 , Amit K. Paul 1
Affiliation
|
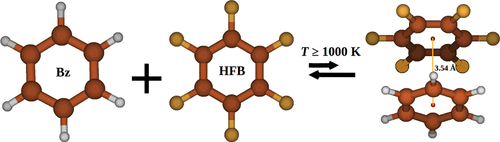
Chemical dynamics simulations are performed to study the association of benzene (Bz) and hexafluorobenzene (HFB) followed by the ensuing dissociation of the Bz–HFB complex. The calculations are done for 1000, 1500, and 2000 K with an impact parameter (b) range of 0–10 Å at each temperature. Almost no complexes are observed to form at b = 8 and 10 Å. Following three different methods of calculation of the temperature-dependent association rate constant kasso(T), the values obtained are 1.67 × 10–10, 1.86 × 10–10, and 2.05 × 10–10 cm3/molecule·s with a standard deviation of approximately 0.1 × 10–10 cm3/molecule·s for T = 1500 K. Among those values of kasso(T), the middle one is obtained by considering a relative translational energy of 3RT/2 at T = 1500 K, and the same is followed to calculate kasso(T) at 1000 and 2000 K. The Arrhenius parameters, using the kasso(T) values at three temperatures, are 0.203 × 10–10 cm3/molecule·s for the pre-exponential factor and −5.79 kcal/mol for the activation energy. The absolute value of the latter is similar to the Bz + HFB association energy of 5.93 kcal/mol. The ensuing dissociation dynamics of the complex is significantly different from the unimolecular dissociation dynamics, and an exponential function fits the N(t – t0)/N(t0) curves comparatively well. The ensuing dissociation is also observed to be independent of time for a statistically large sample size.
中文翻译:

苯-六氟苯分子系统缔合和解离的化学动力学模拟
进行化学动力学模拟以研究苯(Bz)和六氟苯(HFB)的缔合,然后随之解离Bz-HFB配合物。对于1000、1500和2000 K进行了计算,每个温度下的冲击参数(b)范围为0–10Å。在b = 8和10Å处几乎没有观察到络合物的形成。以下温度依赖性结合速率常数计算的三种不同的方法ķ阿索(Ť),所获得的值是1.67×10 -10,1.86×10 -10,和2.05×10 -10厘米3 /分子·S用标准偏差约为0.1×10 –10T = 1500 K时为cm 3 /分子·s 。在k asso(T)的那些值中,中间值是通过考虑在T = 1500 K时的3 RT / 2的相对平移能而得到的,计算ķ ASSO(Ť)在1000和2000 K的阿伦尼乌斯参数,使用ķ阿索(Ť)值在三个温度下,是0.203×10 -10厘米3/分子倍数表示预指数因子,而-5.79 kcal / mol表示活化能。后者的绝对值类似于5.93 kcal / mol的Bz + HFB缔合能。随后的复合物解离动力学与单分子解离动力学显着不同,并且指数函数相对较好地拟合了N(t – t 0)/ N(t 0)曲线。对于统计上大的样本量,随后的解离也与时间无关。
更新日期:2019-05-30
中文翻译:
苯-六氟苯分子系统缔合和解离的化学动力学模拟
进行化学动力学模拟以研究苯(Bz)和六氟苯(HFB)的缔合,然后随之解离Bz-HFB配合物。对于1000、1500和2000 K进行了计算,每个温度下的冲击参数(b)范围为0–10Å。在b = 8和10Å处几乎没有观察到络合物的形成。以下温度依赖性结合速率常数计算的三种不同的方法ķ阿索(Ť),所获得的值是1.67×10 -10,1.86×10 -10,和2.05×10 -10厘米3 /分子·S用标准偏差约为0.1×10 –10T = 1500 K时为cm 3 /分子·s 。在k asso(T)的那些值中,中间值是通过考虑在T = 1500 K时的3 RT / 2的相对平移能而得到的,计算ķ ASSO(Ť)在1000和2000 K的阿伦尼乌斯参数,使用ķ阿索(Ť)值在三个温度下,是0.203×10 -10厘米3/分子倍数表示预指数因子,而-5.79 kcal / mol表示活化能。后者的绝对值类似于5.93 kcal / mol的Bz + HFB缔合能。随后的复合物解离动力学与单分子解离动力学显着不同,并且指数函数相对较好地拟合了N(t – t 0)/ N(t 0)曲线。对于统计上大的样本量,随后的解离也与时间无关。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号