当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
AMOEBA+ Classical Potential for Modeling Molecular Interactions.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-05-28 00:00:00 , DOI: 10.1021/acs.jctc.9b00261 Chengwen Liu 1 , Jean-Philip Piquemal 1, 2, 3 , Pengyu Ren 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-05-28 00:00:00 , DOI: 10.1021/acs.jctc.9b00261 Chengwen Liu 1 , Jean-Philip Piquemal 1, 2, 3 , Pengyu Ren 1
Affiliation
|
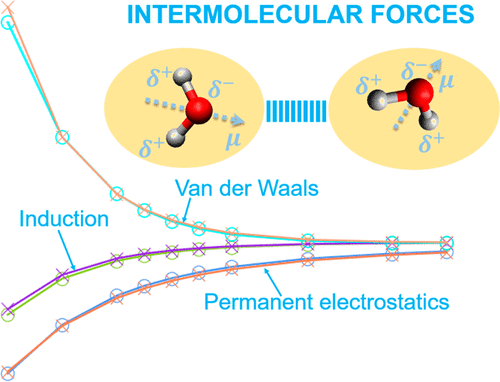
Classical potentials based on isotropic and additive atomic charges have been widely used to model molecules in computers for the past few decades. The crude approximations in the underlying physics are hindering both their accuracy and transferability across chemical and physical environments. Here we present a new classical potential, AMOEBA+, to capture essential intermolecular forces, including permanent electrostatics, repulsion, dispersion, many-body polarization, short-range charge penetration, and charge transfer, by extending the polarizable multipole-based AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications) model. For a set of common organic molecules, we show that AMOEBA+ with general parameters can reproduce both quantum mechanical interactions and energy decompositions according to Symmetry-Adapted Perturbation Theory (SAPT). Additionally, a new water model based on the AMOEBA+ framework captures various liquid-phase properties in molecular dynamics simulations while remaining consistent with SAPT energy decompositions, utilizing both ab initio data and experimental liquid properties. Our results demonstrate that it is possible to improve the physical basis of classical force fields to advance their accuracy and general applicability.
中文翻译:

用于建模分子相互作用的AMOEBA +经典势能。
在过去的几十年中,基于各向同性和加性原子电荷的经典势能已被广泛用于对计算机中的分子进行建模。基础物理学中的粗略近似既阻碍了其准确性,也阻碍了其在化学和物理环境中的可传递性。在这里,我们提出了一种新的经典势能AMOEBA +,它通过扩展可极化的基于多极子的AMOEBA(原子多极子)来捕获基本的分子间力,包括永久性静电,斥力,色散,多体极化,短程电荷渗透和电荷转移。针对生物分子应用的优化能量学模型。对于一组常见的有机分子,我们显示,根据对称自适应扰动理论(SAPT),具有一般参数的AMOEBA +可以重现量子力学相互作用和能量分解。此外,基于AMOEBA +框架的新水模型在分子动力学模拟中捕获了各种液相特性,同时利用从头算数据和实验性液体特性,保持了与SAPT能量分解的一致性。我们的结果表明,可以改善经典力场的物理基础,以提高其准确性和通用性。利用从头算数据和实验液体性质。我们的结果表明,可以改善经典力场的物理基础,以提高其准确性和通用性。利用从头算数据和实验液体性质。我们的结果表明,可以改善经典力场的物理基础,以提高其准确性和通用性。
更新日期:2019-05-28
中文翻译:
用于建模分子相互作用的AMOEBA +经典势能。
在过去的几十年中,基于各向同性和加性原子电荷的经典势能已被广泛用于对计算机中的分子进行建模。基础物理学中的粗略近似既阻碍了其准确性,也阻碍了其在化学和物理环境中的可传递性。在这里,我们提出了一种新的经典势能AMOEBA +,它通过扩展可极化的基于多极子的AMOEBA(原子多极子)来捕获基本的分子间力,包括永久性静电,斥力,色散,多体极化,短程电荷渗透和电荷转移。针对生物分子应用的优化能量学模型。对于一组常见的有机分子,我们显示,根据对称自适应扰动理论(SAPT),具有一般参数的AMOEBA +可以重现量子力学相互作用和能量分解。此外,基于AMOEBA +框架的新水模型在分子动力学模拟中捕获了各种液相特性,同时利用从头算数据和实验性液体特性,保持了与SAPT能量分解的一致性。我们的结果表明,可以改善经典力场的物理基础,以提高其准确性和通用性。利用从头算数据和实验液体性质。我们的结果表明,可以改善经典力场的物理基础,以提高其准确性和通用性。利用从头算数据和实验液体性质。我们的结果表明,可以改善经典力场的物理基础,以提高其准确性和通用性。






























 京公网安备 11010802027423号
京公网安备 11010802027423号