当前位置:
X-MOL 学术
›
Nat. Microbiol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Evaluation of a concatenated protein phylogeny for classification of tailed double-stranded DNA viruses belonging to the order Caudovirales.
Nature Microbiology ( IF 20.5 ) Pub Date : 2019-05-20 , DOI: 10.1038/s41564-019-0448-z Soo Jen Low 1 , Mária Džunková 1 , Pierre-Alain Chaumeil 1 , Donovan H Parks 1 , Philip Hugenholtz 1
Nature Microbiology ( IF 20.5 ) Pub Date : 2019-05-20 , DOI: 10.1038/s41564-019-0448-z Soo Jen Low 1 , Mária Džunková 1 , Pierre-Alain Chaumeil 1 , Donovan H Parks 1 , Philip Hugenholtz 1
Affiliation
|
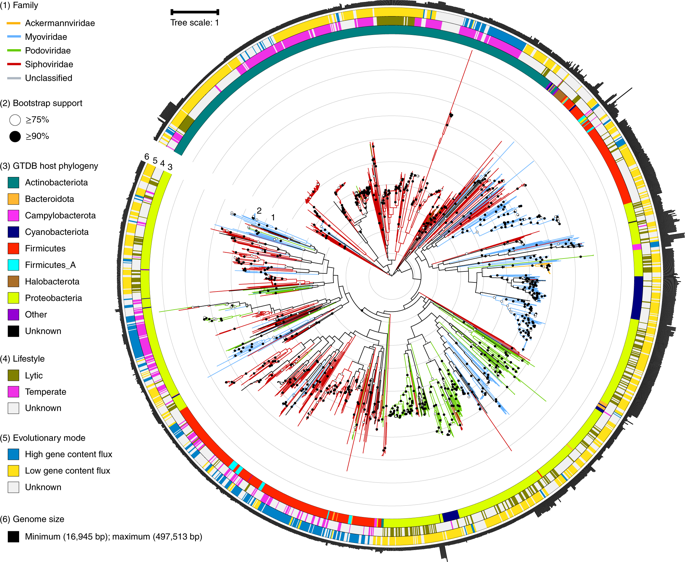
Viruses of bacteria and archaea are important players in global carbon cycling as well as drivers of host evolution, yet the taxonomic classification of viruses remains a challenge due to their genetic diversity and absence of universally conserved genes. Traditional classification approaches employ a combination of phenotypic and genetic information which is no longer scalable in the era of bulk viral genome recovery through metagenomics. Here, we evaluate a phylogenetic approach for the classification of tailed double-stranded DNA viruses from the order Caudovirales by inferring a phylogeny from the concatenation of 77 single-copy protein markers using a maximum-likelihood method. Our approach is largely consistent with the International Committee on Taxonomy of Viruses, with 72 and 89% congruence at the subfamily and genus levels, respectively. Discrepancies could be attributed to misclassifications and a small number of highly mosaic genera confounding the phylogenetic signal. We also show that confidently resolved nodes in the concatenated protein tree are highly reproducible across different software and models, and conclude that the approach can serve as a framework for a rank-normalized taxonomy of most tailed double-stranded DNA viruses.
中文翻译:

评估级联蛋白质的系统发育,用于分类属于尾病毒科的有尾双链DNA病毒。
细菌和古细菌的病毒是全球碳循环的重要参与者,也是宿主进化的驱动力,但是由于其遗传多样性和缺乏普遍保守的基因,病毒的分类学分类仍然是一个挑战。传统的分类方法采用了表型和遗传信息的组合,在通过宏基因组学进行大规模病毒基因组回收的时代,这种信息已不再可扩展。在这里,我们通过使用最大似然法从77个单拷贝蛋白质标记的串联中推断出系统发育,从而评估了Caudovirales尾部双链DNA病毒分类的系统发育方法。我们的方法在很大程度上与国际病毒分类学委员会一致,在亚科和属水平上分别达到72%和89%的一致性。差异可能归因于分类错误和少数高度镶嵌的属混淆了系统发育信号。我们还显示,在不同软件和模型之间,连接蛋白质树中的可信解析结点具有很高的可重现性,并得出结论,该方法可以用作大多数尾部双链DNA病毒的秩归一化分类法的框架。
更新日期:2019-05-21
中文翻译:
评估级联蛋白质的系统发育,用于分类属于尾病毒科的有尾双链DNA病毒。
细菌和古细菌的病毒是全球碳循环的重要参与者,也是宿主进化的驱动力,但是由于其遗传多样性和缺乏普遍保守的基因,病毒的分类学分类仍然是一个挑战。传统的分类方法采用了表型和遗传信息的组合,在通过宏基因组学进行大规模病毒基因组回收的时代,这种信息已不再可扩展。在这里,我们通过使用最大似然法从77个单拷贝蛋白质标记的串联中推断出系统发育,从而评估了Caudovirales尾部双链DNA病毒分类的系统发育方法。我们的方法在很大程度上与国际病毒分类学委员会一致,在亚科和属水平上分别达到72%和89%的一致性。差异可能归因于分类错误和少数高度镶嵌的属混淆了系统发育信号。我们还显示,在不同软件和模型之间,连接蛋白质树中的可信解析结点具有很高的可重现性,并得出结论,该方法可以用作大多数尾部双链DNA病毒的秩归一化分类法的框架。




























 京公网安备 11010802027423号
京公网安备 11010802027423号