当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Hierarchical Approach to Predict Conformation-Dependent Histidine Protonation States in Stable and Flexible Proteins.
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2019-06-07 , DOI: 10.1021/acs.jpcb.9b00656 Serzhan N Sakipov 1 , Jose C Flores-Canales 1 , Maria G Kurnikova 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2019-06-07 , DOI: 10.1021/acs.jpcb.9b00656 Serzhan N Sakipov 1 , Jose C Flores-Canales 1 , Maria G Kurnikova 1
Affiliation
|
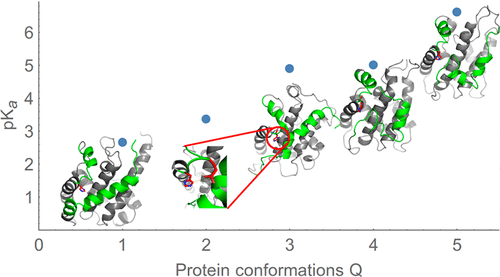
Solution acidity measured by pH is an important environmental factor that affects protein structure. It influences the protonation state of protein residues, which in turn may be coupled to protein conformational changes, unfolding, and ligand binding. It remains difficult to compute and measure the p Ka of individual residues, as well as to relate them to pH-dependent protein transitions. This paper presents a hierarchical approach to compute the p Ka of individual protonatable residues, specifically histidines, coupled with underlying structural changes of a protein. A fast and efficient free energy perturbation (FEP) algorithm has also been developed utilizing a fast implementation of standard molecular dynamics (MD) algorithms. Specifically, a CUDA version of the AMBER MD engine is used in this paper. Eight histidine p Ka's are computed in a diverse set of pH stable proteins to demonstrate the proposed approach's utility and assess the predictive quality of the AMBER FF99SB force field. A reference molecule is carefully selected and tested for convergence. A hierarchical approach is used to model p Ka's of the six histidine residues of the diphtheria toxin translocation domain (DTT), which exhibits a diverse ensemble of individual conformations and pH-dependent unfolding. The hierarchical approach consists of first sampling equilibrium conformational ensembles of a protein with protonated and neutral histidine residues via long equilibrium MD simulations (Flores-Canales, J. C.; et al. bioRxiv, 2019, 572040). A clustering method is then used to identify sampled protein conformations, and p Ka's of histidines in each protein conformation are computed. Finally, an ensemble averaging formalism is developed to compute weighted average histidine p Ka's. These can be compared with an apparent experimentally measured p Ka of the DTT protein and thus allows us to propose a mechanism of pH-dependent unfolding of the DTT protein.
中文翻译:

预测稳定和灵活蛋白质中依赖构象的组氨酸质子化状态的分层方法。
通过pH测量的溶液酸度是影响蛋白质结构的重要环境因素。它影响蛋白质残基的质子化状态,进而可能与蛋白质构象变化,展开和配体结合相关。仍然难以计算和测量单个残基的p Ka,以及将其与pH依赖的蛋白质跃迁相关联。本文提出了一种分层方法来计算单个可质子化残基(特别是组氨酸)的p Ka,以及蛋白质的潜在结构变化。利用标准分子动力学(MD)算法的快速实现,还开发了一种快速有效的自由能扰动(FEP)算法。具体来说,本文使用的是AMBER MD引擎的CUDA版本。八个组氨酸p Ka' 在各种pH稳定蛋白中计算s,以证明所提出的方法的实用性并评估AMBER FF99SB力场的预测质量。仔细选择参考分子并测试其收敛性。使用分级方法来模拟白喉毒素易位结构域(DTT)的六个组氨酸残基的p Ka值,该值显示出各种单独的构象和pH依赖的展开。分层方法包括通过长时间的平衡MD模拟(Flores-Canales,JC; et al.bioRxiv,2019,572040),首先对具有质子化和中性组氨酸残基的蛋白质进行平衡构象采样。然后使用聚类方法来识别采样的蛋白质构象,并计算每种蛋白质构象中的组氨酸的p Ka值。最终,开发了一种综合平均形式,以计算加权平均组氨酸p Ka值。可以将它们与DTT蛋白的表观实验测量值p Ka相比较,从而使我们能够提出DTT蛋白的pH依赖性解折叠机制。
更新日期:2019-06-07
中文翻译:
预测稳定和灵活蛋白质中依赖构象的组氨酸质子化状态的分层方法。
通过pH测量的溶液酸度是影响蛋白质结构的重要环境因素。它影响蛋白质残基的质子化状态,进而可能与蛋白质构象变化,展开和配体结合相关。仍然难以计算和测量单个残基的p Ka,以及将其与pH依赖的蛋白质跃迁相关联。本文提出了一种分层方法来计算单个可质子化残基(特别是组氨酸)的p Ka,以及蛋白质的潜在结构变化。利用标准分子动力学(MD)算法的快速实现,还开发了一种快速有效的自由能扰动(FEP)算法。具体来说,本文使用的是AMBER MD引擎的CUDA版本。八个组氨酸p Ka' 在各种pH稳定蛋白中计算s,以证明所提出的方法的实用性并评估AMBER FF99SB力场的预测质量。仔细选择参考分子并测试其收敛性。使用分级方法来模拟白喉毒素易位结构域(DTT)的六个组氨酸残基的p Ka值,该值显示出各种单独的构象和pH依赖的展开。分层方法包括通过长时间的平衡MD模拟(Flores-Canales,JC; et al.bioRxiv,2019,572040),首先对具有质子化和中性组氨酸残基的蛋白质进行平衡构象采样。然后使用聚类方法来识别采样的蛋白质构象,并计算每种蛋白质构象中的组氨酸的p Ka值。最终,开发了一种综合平均形式,以计算加权平均组氨酸p Ka值。可以将它们与DTT蛋白的表观实验测量值p Ka相比较,从而使我们能够提出DTT蛋白的pH依赖性解折叠机制。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号