当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Probing the high potency of pyrazolyl pyrimidinetriones and thioxopyrimidinediones as selective and efficient non-nucleotide inhibitors of recombinant human ectonucleotidases.
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2019-04-02 , DOI: 10.1016/j.bioorg.2019.03.067 Hina Andleeb 1 , Shahid Hameed 2 , Syeda Abida Ejaz 3 , Imtiaz Khan 4 , Sumera Zaib 3 , Joanna Lecka 5 , Jean Sévigny 5 , Jamshed Iqbal 3
Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2019-04-02 , DOI: 10.1016/j.bioorg.2019.03.067 Hina Andleeb 1 , Shahid Hameed 2 , Syeda Abida Ejaz 3 , Imtiaz Khan 4 , Sumera Zaib 3 , Joanna Lecka 5 , Jean Sévigny 5 , Jamshed Iqbal 3
Affiliation
|
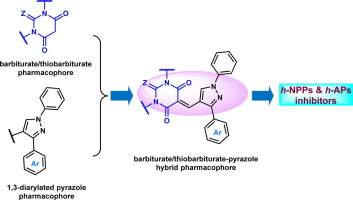
With the aim to discover novel, efficient and selective inhibitors of human alkaline phosphatase and nucleotide pyrophosphatase enzymes, two new series of pyrazolyl pyrimidinetriones (PPTs) (6a-g) and thioxopyrimidinediones (PTPs) (6h-n) were synthesized in good chemical yields using Knoevenagel condensation reaction between pyrazole carbaldehydes (4a-g) and pharmacologically active N-alkylated pyrimidinetrione (5a) and thioxopyrimidinedione (5b). The inhibition potential of the synthesized hybrid compounds was evaluated against human alkaline phosphatase (h-TNAP and h-IAP) and ectonucleotidase (h-NPP1 and h-NPP3) enzymes. Most of the tested analogs were highly potent with a variable degree of inhibition depending on the functionalized hybrid structure. The detailed structure-activity relationship (SAR) of PPT and PTP derivatives suggested that the compound with unsubstituted phenyl ring from PPT series led to selective and potent inhibition (6a; IC50 = 0.33 ± 0.02 µM) of h-TNAP, whereas compound 6c selectively inhibited h-IAP isozyme with IC50 value of 0.86 ± 0.04 µM. Similarly, compounds 6b and 6h were identified as the lead scaffolds against h-NPP1 and h-NPP3, respectively. The probable binding modes for the most potent inhibitors were elucidated through molecular docking analysis. Structure-activity relationships, mechanism of action, cytotoxic effects and druglikeness properties are also discussed.
中文翻译:

探究吡唑基嘧啶三酮和硫代嘧啶二酮作为重组人外切核苷酸酶的选择性和有效非核苷酸抑制剂的高效力。
为了发现新型,有效和选择性的人类碱性磷酸酶和核苷酸焦磷酸酶抑制剂,合成了两个新的吡唑基嘧啶三酮(PPT)(6a-g)和硫代嘧啶二酮(PTP)(6h-n),其化学收率很高。使用吡唑甲醛(4a-g)与药理活性N-烷基化嘧啶三酮(5a)和硫代嘧啶二酮(5b)之间的Knoevenagel缩合反应。评估了合成杂合化合物对人碱性磷酸酶(h-TNAP和h-IAP)和胞外核苷酸酶(h-NPP1和h-NPP3)的抑制潜力。取决于功能化的杂合结构,大多数测试的类似物都是高度有效的,具有不同程度的抑制作用。PPT和PTP衍生物的详细结构-活性关系(SAR)表明,具有PPT系列未取代苯环的化合物可导致对h-TNAP的选择性强效抑制(6a; IC50 = 0.33±0.02 µM),而化合物6c可选择性抑制h-TNAP抑制h-IAP同工酶,IC50值为0.86±0.04 µM。同样,化合物6b和6h被分别确定为对抗h-NPP1和h-NPP3的铅支架。通过分子对接分析阐明了最有效抑制剂的可能结合方式。还讨论了构效关系,作用机理,细胞毒性作用和药物相似性。而化合物6c选择性抑制h-IAP同工酶,IC50值为0.86±0.04 µM。同样,化合物6b和6h被分别确定为对抗h-NPP1和h-NPP3的铅支架。通过分子对接分析阐明了最有效抑制剂的可能结合方式。还讨论了构效关系,作用机理,细胞毒性作用和药物相似性。而化合物6c选择性抑制h-IAP同工酶,IC50值为0.86±0.04 µM。同样,化合物6b和6h被分别确定为对抗h-NPP1和h-NPP3的铅支架。通过分子对接分析阐明了最有效抑制剂的可能结合方式。还讨论了构效关系,作用机理,细胞毒性作用和药物相似性。
更新日期:2019-04-02
中文翻译:
探究吡唑基嘧啶三酮和硫代嘧啶二酮作为重组人外切核苷酸酶的选择性和有效非核苷酸抑制剂的高效力。
为了发现新型,有效和选择性的人类碱性磷酸酶和核苷酸焦磷酸酶抑制剂,合成了两个新的吡唑基嘧啶三酮(PPT)(6a-g)和硫代嘧啶二酮(PTP)(6h-n),其化学收率很高。使用吡唑甲醛(4a-g)与药理活性N-烷基化嘧啶三酮(5a)和硫代嘧啶二酮(5b)之间的Knoevenagel缩合反应。评估了合成杂合化合物对人碱性磷酸酶(h-TNAP和h-IAP)和胞外核苷酸酶(h-NPP1和h-NPP3)的抑制潜力。取决于功能化的杂合结构,大多数测试的类似物都是高度有效的,具有不同程度的抑制作用。PPT和PTP衍生物的详细结构-活性关系(SAR)表明,具有PPT系列未取代苯环的化合物可导致对h-TNAP的选择性强效抑制(6a; IC50 = 0.33±0.02 µM),而化合物6c可选择性抑制h-TNAP抑制h-IAP同工酶,IC50值为0.86±0.04 µM。同样,化合物6b和6h被分别确定为对抗h-NPP1和h-NPP3的铅支架。通过分子对接分析阐明了最有效抑制剂的可能结合方式。还讨论了构效关系,作用机理,细胞毒性作用和药物相似性。而化合物6c选择性抑制h-IAP同工酶,IC50值为0.86±0.04 µM。同样,化合物6b和6h被分别确定为对抗h-NPP1和h-NPP3的铅支架。通过分子对接分析阐明了最有效抑制剂的可能结合方式。还讨论了构效关系,作用机理,细胞毒性作用和药物相似性。而化合物6c选择性抑制h-IAP同工酶,IC50值为0.86±0.04 µM。同样,化合物6b和6h被分别确定为对抗h-NPP1和h-NPP3的铅支架。通过分子对接分析阐明了最有效抑制剂的可能结合方式。还讨论了构效关系,作用机理,细胞毒性作用和药物相似性。
































 京公网安备 11010802027423号
京公网安备 11010802027423号