当前位置:
X-MOL 学术
›
Batteries Supercaps
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanism of Lithium Cation Hopping between Tetragonal Thiophene Cages
Batteries & Supercaps ( IF 5.1 ) Pub Date : 2019-04-25 , DOI: 10.1002/batt.201900043 Pouya Partovi‐Azar 1 , Daniel Sebastiani 1
Batteries & Supercaps ( IF 5.1 ) Pub Date : 2019-04-25 , DOI: 10.1002/batt.201900043 Pouya Partovi‐Azar 1 , Daniel Sebastiani 1
Affiliation
|
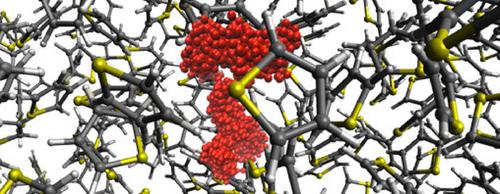
We report on the atomistic mechanism of elementary hopping processes of Li+ ions in liquid thiophene obtained from ab‐initio molecular dynamics simulations. We observe the formation of cage structures which solvate the cation. Besides the actual molecular solvation structure, we provide an analysis of the pathway and timescale of basic Li+ diffusion steps in terms of coordination by sulfur atoms. We compare our results to the situation in a thiophene derivative, namely 3,4‐ethylenedioxythiophene (EDOT). The calculations reveal that in both thiophene and EDOT liquids, a tetrahedral structure is formed around the Li+ ion. While in the case of the former, the Li cation is coordinated by four sulfur atoms, in the latter case it is surrounded by four oxygens. The tetrahedrons act as cages, which accommodate the cation for a considerable duration (of the order of 100 ps). The elementary diffusion step occurs through a “permeable edge” of the tetrahedron formed by two sulfur (or oxygen) atoms at a characteristic distance. This finding indicates that Li+ conduction in thiophene derivatives can be improved by rationally designing the compound in such a way that maximizes the occurrence of sulfur atoms at that particular distance from each other.
中文翻译:

四方噻吩笼之间锂阳离子跳跃的机理
我们报告了从头算分子动力学模拟获得的液态噻吩中Li +离子基本跳跃过程的原子机理。我们观察到溶剂化阳离子的笼结构的形成。除了实际的分子溶剂化结构外,我们还通过硫原子的配位分析了基本的Li +扩散步骤的路径和时标。我们将结果与噻吩衍生物3,4-乙烯二氧噻吩(EDOT)的情况进行了比较。计算表明,在噻吩和EDOT液体中,Li +周围均形成四面体结构离子。在前者的情况下,Li阳离子由四个硫原子配位,在后者的情况下,它被四个氧包围。四面体充当笼子,可以在相当长的时间内(约100 ps)容纳阳离子。基本扩散步骤通过由特征距离处的两个硫(或氧)原子形成的四面体的“可渗透边缘”发生。该发现表明,通过合理地设计化合物以最大程度地增加在彼此之间的该特定距离处硫原子的出现,可以改善噻吩衍生物中的Li +传导性。
更新日期:2019-04-25
中文翻译:
四方噻吩笼之间锂阳离子跳跃的机理
我们报告了从头算分子动力学模拟获得的液态噻吩中Li +离子基本跳跃过程的原子机理。我们观察到溶剂化阳离子的笼结构的形成。除了实际的分子溶剂化结构外,我们还通过硫原子的配位分析了基本的Li +扩散步骤的路径和时标。我们将结果与噻吩衍生物3,4-乙烯二氧噻吩(EDOT)的情况进行了比较。计算表明,在噻吩和EDOT液体中,Li +周围均形成四面体结构离子。在前者的情况下,Li阳离子由四个硫原子配位,在后者的情况下,它被四个氧包围。四面体充当笼子,可以在相当长的时间内(约100 ps)容纳阳离子。基本扩散步骤通过由特征距离处的两个硫(或氧)原子形成的四面体的“可渗透边缘”发生。该发现表明,通过合理地设计化合物以最大程度地增加在彼此之间的该特定距离处硫原子的出现,可以改善噻吩衍生物中的Li +传导性。
































 京公网安备 11010802027423号
京公网安备 11010802027423号