当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
H-最优化在二酮吡咯并吡咯中分子间相互作用和电荷转移积分中的作用
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-03-21 00:00:00 , DOI: 10.1021/acs.jpca.9b01275
Jesus Calvo-Castro 1 , Alan R. Kennedy 2 , Callum J. McHugh 3
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-03-21 00:00:00 , DOI: 10.1021/acs.jpca.9b01275
Jesus Calvo-Castro 1 , Alan R. Kennedy 2 , Callum J. McHugh 3
Affiliation
|
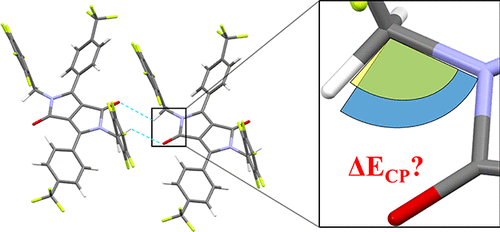
迫切需要小型有机共轭体系,其在固态下显示一维堆积图案,从而促进电荷传播。非共价相互作用虽然微弱,但可以协同地为那些超分子结构提供大的结合能和相关的热完整性。在众多有助于整体晶格能量和电荷传播超分子体系结构稳定性的分子间相互作用中,众所周知,氢键相互作用起着举足轻重的作用。尽管做出了关键性的贡献,但X射线晶体学数据中氢原子的位置却被参数化,这可能导致计算出的分子间相互作用发生显着变化。在此处,我们首次报告了对X射线结构中H原子的优化在二酮吡咯并吡咯(DPPs)分子间相互作用能的计算中的作用的分析。使用了一个大型数据集,该数据集包含来自19个不同的基于DPP的系统的94个二聚体对,其中包括三个颜料类似物。总体上,考虑使用并在6-311G(d)水平使用M06-2X密度官能团优化了超过1400个H–X化学键。计算了H优化的几何结构的分子间相互作用,并将其与未优化的几何结构进行了比较。我们报告说,在研究的94个二聚体对中有35个(37%)中,计算的分子间相互作用至少为2.5 kJ mol。使用了一个大型数据集,该数据集包含来自19个不同的基于DPP的系统的94个二聚体对,其中包括三个颜料类似物。总体上,考虑使用并在6-311G(d)水平使用M06-2X密度官能团优化了超过1400个H–X化学键。计算了H优化的几何结构的分子间相互作用,并将其与未优化的几何结构进行了比较。我们报告说,在研究的94个二聚体对中有35个(37%)中,计算的分子间相互作用至少为2.5 kJ mol。使用了一个大型数据集,该数据集包含来自19个不同的基于DPP的系统的94个二聚体对,其中包括三个颜料类似物。总体上,考虑使用并在6-311G(d)水平使用M06-2X密度官能团优化了超过1400个H–X化学键。计算了H优化的几何结构的分子间相互作用,并将其与未优化的几何结构进行了比较。我们报告说,在研究的94个二聚体对中有35个(37%)中,计算的分子间相互作用至少为2.5 kJ mol。计算了H优化的几何结构的分子间相互作用,并将其与未优化的几何结构进行了比较。我们报告说,在研究的94个二聚体对中有35个(37%)中,计算的分子间相互作用至少为2.5 kJ mol。计算了H优化的几何结构的分子间相互作用,并将其与未优化的几何结构进行了比较。我们报告说,在研究的94个二聚体对中有35个(37%)中,计算的分子间相互作用至少为2.5 kJ mol。到H优化的几何图形时,增大–1。反过来,对94个二聚体对中的8个(8%)进行H优化后,得出的计算值较低,其中一种情况显示的差异大于2.5 kJ mol –1。由于电子密度的变化可忽略不计,并且波函数重叠,计算出的空穴和电子的传递积分变化始终低于1 kJ mol –1。在确定超分子结构和电荷传播通道的热完整性时,观察到的分子间相互作用的变化起着至关重要的作用,因此,在没有中子衍射数据的情况下,应在计算之前优化H原子。我们设想本文的结果将引起广泛的科学界的兴趣,该界致力于了解有机共轭体系中的分子间相互作用以及实现优异的电荷转移介导材料,并且鉴于研究的分子间相互作用过多,结果是不仅仅限于基于DPP的体系结构。

"点击查看英文标题和摘要"
更新日期:2019-03-21
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号