当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Effect of aluminum vacancies on the H2O2 or H2O interaction with a gamma‐AlOOH surface. A solid‐state DFT study
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2019-02-20 , DOI: 10.1002/qua.25920 Alexander G. Medvedev 1, 2 , Alexey A. Mikhaylov 1, 2 , Ivan Yu. Chernyshov 1, 3 , Mikhail V. Vener 3 , Ovadia Lev 4 , Petr V. Prikhodchenko 1
International Journal of Quantum Chemistry ( IF 2.3 ) Pub Date : 2019-02-20 , DOI: 10.1002/qua.25920 Alexander G. Medvedev 1, 2 , Alexey A. Mikhaylov 1, 2 , Ivan Yu. Chernyshov 1, 3 , Mikhail V. Vener 3 , Ovadia Lev 4 , Petr V. Prikhodchenko 1
Affiliation
|
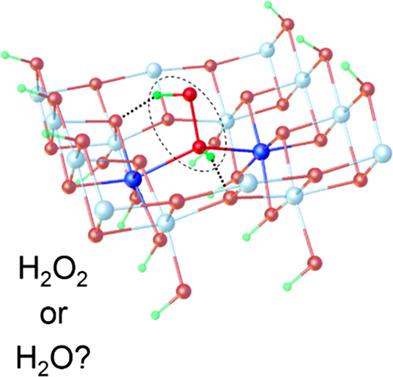
The adsorption of a single H2O2 or H2O molecule on a family of periodic slab models of γ‐AlOOH is studied by solid‐state DFT. The single H2O2 or Н2О molecule interacts with the perfect (010) slab by intermolecular hydrogen bonds (H‐bonds). In the models of γ‐AlOOH with oxygen and aluminum vacancies, H2O2 or Н2О also forms covalent O∙∙∙Al bonds. The energies of covalent O∙∙∙Al and H‐bonds are estimated by a combined approach based on simultaneous consideration of the total binding energies with BSSE correction and empirical schemes of the Н‐bond energy evaluation. The O∙∙∙Al bond energy ranges from ~75 to ~156 kJ mol−1. The total energy of H‐bond interactions in the case of H2O2 exceeds that of Н2О by ~30 kJ mol−1 for all considered slab models. In contrast to Н2О, a H2O2 molecule always forms two H‐bonds as the proton donor. The energy of these bonds noticeably increase on defect γ‐AlOOH surfaces in comparison with the perfect slab due to formation of short (strong) H‐bonds by adsorbed H2O2.
中文翻译:

铝空位对H2O2或H2O与γ-AlOOH表面相互作用的影响。固态DFT研究
通过固态DFT研究了单个H 2 O 2或H 2 O分子在γ-AlOOH周期平板模型族上的吸附。单ħ 2 ö 2或Н 2 О分子相互作用与由分子间氢键(H-键)的完美(010)板。在γ-AlOOH的与氧和铝空位,H的模型2 Ò 2或Н 2О还形成O∙∙∙Al共价键。共价O∙∙∙Al和H键的能量是通过结合方法同时估算总结合能和BSSE校正以及Н键能量评估的经验方案来估算的。O∙∙∙Al的键能范围为〜75至〜156 kJ mol -1。氢键相互作用的在H的情况下的总能量2 ö 2超过Н的2 О由〜30千焦耳摩尔-1对于所有考虑的板坯的模型。在对比Н 2 О,一个ħ 2 ö 2分子总是形成两个氢键作为质子供体。与完美平板相比,缺陷γ-AlOOH表面上这些键的能量显着增加,这是由于吸附的H 2 O 2形成了短(强)氢键。
更新日期:2019-02-20
中文翻译:
铝空位对H2O2或H2O与γ-AlOOH表面相互作用的影响。固态DFT研究
通过固态DFT研究了单个H 2 O 2或H 2 O分子在γ-AlOOH周期平板模型族上的吸附。单ħ 2 ö 2或Н 2 О分子相互作用与由分子间氢键(H-键)的完美(010)板。在γ-AlOOH的与氧和铝空位,H的模型2 Ò 2或Н 2О还形成O∙∙∙Al共价键。共价O∙∙∙Al和H键的能量是通过结合方法同时估算总结合能和BSSE校正以及Н键能量评估的经验方案来估算的。O∙∙∙Al的键能范围为〜75至〜156 kJ mol -1。氢键相互作用的在H的情况下的总能量2 ö 2超过Н的2 О由〜30千焦耳摩尔-1对于所有考虑的板坯的模型。在对比Н 2 О,一个ħ 2 ö 2分子总是形成两个氢键作为质子供体。与完美平板相比,缺陷γ-AlOOH表面上这些键的能量显着增加,这是由于吸附的H 2 O 2形成了短(强)氢键。
































 京公网安备 11010802027423号
京公网安备 11010802027423号