Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Se–C Cleavage of Hexane Selenol at Steps on Au(111)
Langmuir ( IF 3.7 ) Pub Date : 2018-02-15 00:00:00 , DOI: 10.1021/acs.langmuir.7b03713 Zahra Besharat 1, 2 , Milad Ghadami Yazdi 1 , Deborah Wakeham 2 , Magnus Johnson 2 , Mark W. Rutland 2, 3 , Mats Göthelid 1 , Henrik Grönbeck 4
Langmuir ( IF 3.7 ) Pub Date : 2018-02-15 00:00:00 , DOI: 10.1021/acs.langmuir.7b03713 Zahra Besharat 1, 2 , Milad Ghadami Yazdi 1 , Deborah Wakeham 2 , Magnus Johnson 2 , Mark W. Rutland 2, 3 , Mats Göthelid 1 , Henrik Grönbeck 4
Affiliation
|
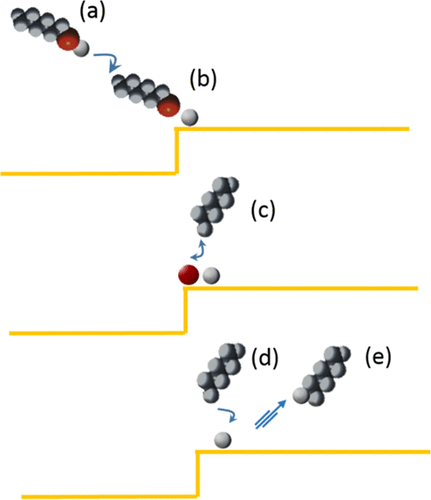
Selenols are considered as an alternative to thiols in self-assembled monolayers, but the Se–C bond is one limiting factor for their usefulness. In this study, we address the stability of the Se–C bond by a combined experimental and theoretical investigation of gas-phase-deposited hexane selenol (CH3(CH2)5SeH) on Au(111) using photoelectron spectroscopy, scanning tunneling microscopy, and density functional theory (DFT). Experimentally, we find that initial adsorption leaves atomic Se on the surface without any carbon left on the surface, whereas further adsorption generates a saturated selenolate layer. The Se 3d component from atomic Se appears at 0.85 eV lower binding energy than the selenolate-related component. DFT calculations show that the most stable structure of selenols on Au(111) is in the form of RSe–Au–SeR complexes adsorbed on the unreconstructed Au(111) surface. This is similar to thiols on Au(111). Calculated Se 3d core-level shifts between elemental Se and selenolate in this structure nicely reproduce the experimentally recorded shifts. Dissociation of RSeH and subsequent formation of RH are found to proceed with high barriers on defect-free Au(111) terraces, with the highest barrier for scissoring R–Se. However, at steps, these barriers are considerably lower, allowing for Se–C bond breaking and hexane desorption, leaving elemental Se at the surface. Hexane is formed by replacing the Se–C bond with a H–C bond by using the hydrogen liberated from the selenol to selenolate transformation.
中文翻译:

在Au(111)台阶上对Selenol的Se-C裂解
硒醇被认为是自组装单分子层中硫醇的替代物,但Se–C键是其实用性的限制因素。在这项研究中,我们通过气相沉积己烷硒醇(CH 3(CH 2)5)的实验和理论研究相结合的方法,解决了Se-C键的稳定性SeH)在Au(111)上使用光电子能谱,扫描隧道显微镜和密度泛函理论(DFT)。通过实验,我们发现初始吸附在表面上留下了原子Se,而表面上没有任何碳,而进一步的吸附会产生饱和的硒酸盐层。来自硒原子的硒3d组分的结合能比硒酸酯相关组分低0.85 eV。DFT计算表明,Au(111)上硒醇的最稳定结构是吸附在未重建的Au(111)表面上的RSe–Au–SeR络合物的形式。这类似于Au(111)上的硫醇。在该结构中,元素Se和硒代硒酸盐之间计算的Se 3d核心能级位移很好地再现了实验记录的位移。发现RSeH的解离和随后的RH的形成在无缺陷的Au(111)平台上具有高势垒,而对R-Se的剪切势垒最高。但是,在台阶处,这些势垒要低得多,允许Se-C键断裂和己烷解吸,从而在表面留下元素Se。己烷是通过使用从硒醇转化为硒酸酯的氢取代的HC键取代Se-C键而形成的。
更新日期:2018-02-15
中文翻译:
在Au(111)台阶上对Selenol的Se-C裂解
硒醇被认为是自组装单分子层中硫醇的替代物,但Se–C键是其实用性的限制因素。在这项研究中,我们通过气相沉积己烷硒醇(CH 3(CH 2)5)的实验和理论研究相结合的方法,解决了Se-C键的稳定性SeH)在Au(111)上使用光电子能谱,扫描隧道显微镜和密度泛函理论(DFT)。通过实验,我们发现初始吸附在表面上留下了原子Se,而表面上没有任何碳,而进一步的吸附会产生饱和的硒酸盐层。来自硒原子的硒3d组分的结合能比硒酸酯相关组分低0.85 eV。DFT计算表明,Au(111)上硒醇的最稳定结构是吸附在未重建的Au(111)表面上的RSe–Au–SeR络合物的形式。这类似于Au(111)上的硫醇。在该结构中,元素Se和硒代硒酸盐之间计算的Se 3d核心能级位移很好地再现了实验记录的位移。发现RSeH的解离和随后的RH的形成在无缺陷的Au(111)平台上具有高势垒,而对R-Se的剪切势垒最高。但是,在台阶处,这些势垒要低得多,允许Se-C键断裂和己烷解吸,从而在表面留下元素Se。己烷是通过使用从硒醇转化为硒酸酯的氢取代的HC键取代Se-C键而形成的。
































 京公网安备 11010802027423号
京公网安备 11010802027423号