当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A Multireference Ab Initio Study of the Diradical Isomers of Pyrazine
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-02-19 00:00:00 , DOI: 10.1021/acs.jpca.8b12440 Thais Scott 1, 2 , Reed Nieman 3 , Adam Luxon 1 , Boyi Zhang 1 , Hans Lischka 3, 4 , Laura Gagliardi 2 , Carol A. Parish 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-02-19 00:00:00 , DOI: 10.1021/acs.jpca.8b12440 Thais Scott 1, 2 , Reed Nieman 3 , Adam Luxon 1 , Boyi Zhang 1 , Hans Lischka 3, 4 , Laura Gagliardi 2 , Carol A. Parish 1
Affiliation
|
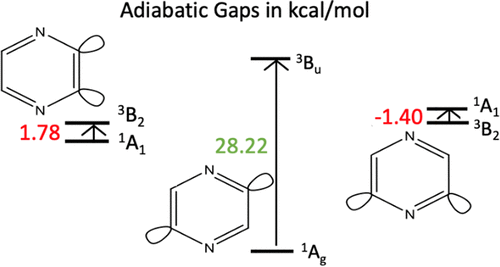
Three diradical pyrazine isomers were characterized using highly correlated, multireference methods. The lowest lying singlet and triplet state geometries of 2,3-didehydropyrazine (ortho), 2,5-didehydropyrazine (para), and 2,6-didehydropyrazine (meta) were determined. Two active reference spaces were utilized. The complete active space (CAS) (8,8) includes the σ and σ* orbitals on the dehydrocarbon atoms as well as the valence π and π* orbitals. The CAS (12,10) reference space includes two additional orbitals corresponding to the in-phase and out-of-phase nitrogen lone pair orbitals. Adiabatic and vertical gaps between the lowest lying singlet and triplet states, optimized geometries, canonicalized orbital energies, unpaired electron densities, and spin polarization effects were compared. We find that the singlet states of each diradical isomer contain two significantly weighted configurations, and the larger active space is necessary for the proper physical characterization of both the singlet and triplet states. The singlet–triplet splitting is very small for the 2,3-didehydropyrazine (ortho) and 2,6-didehydropyrazine (meta) isomers (+1.8 and −1.4 kcal/mol, respectively) and significant for the 2,5-didehydropyrazine (para) isomer (+28.2 kcal/mol). Singlet geometries show through-space interactions between the dehydocarbon atoms in the 2,3-didehydropyrazine (ortho) and 2,6-didehydropyrazine (meta) isomers. An analysis of the effectively unpaired electrons suggests that the 2,5-didehydropyrazine (para) isomer also displays through-bond coupling between the diradical electrons.
中文翻译:

吡嗪双自由基异构体的多参考从头算研究
使用高度相关的多参考方法对三种双自由基吡嗪异构体进行了表征。2,3-didehydropyrazine(邻),2,5-didehydropyrazine(对位)和2,6-didehydropyrazine(间位)的最低单线态和三线态几何)确定。利用了两个有效的参考空间。完整的活性空间(CAS)(8,8)包括脱碳原子上的σ和σ*轨道以及价和π和π*轨道。CAS(12,10)参考空间包括对应于同相和异相氮孤对轨道的两个附加轨道。比较了最低单线态和三线态之间的绝热和垂直间隙,优化的几何形状,规范化的轨道能量,不成对的电子密度以及自旋极化效应。我们发现,每个双自由基异构体的单重态包含两个显着加权的构型,并且较大的活性空间对于正确地表征单重态和三重态都必不可少。2,3-二氢吡嗪的单重态-三重态分裂很小(ortho)和2,6-didehydropyrazine(meta)异构体(分别为+1.8和-1.4 kcal / mol),对于2,5-didehydropyrazine(para)异构体(+28.2 kcal / mol)显着。单重态的几何形状显示了2,3-二氢吡嗪(邻位)和2,6-二氢吡嗪(间位)异构体中的脱氢碳原子之间的全空间相互作用。对有效未成对电子的分析表明,2,5-二氢吡嗪(对)异构体在双自由基电子之间也显示出键间耦合。
更新日期:2019-02-19
中文翻译:
吡嗪双自由基异构体的多参考从头算研究
使用高度相关的多参考方法对三种双自由基吡嗪异构体进行了表征。2,3-didehydropyrazine(邻),2,5-didehydropyrazine(对位)和2,6-didehydropyrazine(间位)的最低单线态和三线态几何)确定。利用了两个有效的参考空间。完整的活性空间(CAS)(8,8)包括脱碳原子上的σ和σ*轨道以及价和π和π*轨道。CAS(12,10)参考空间包括对应于同相和异相氮孤对轨道的两个附加轨道。比较了最低单线态和三线态之间的绝热和垂直间隙,优化的几何形状,规范化的轨道能量,不成对的电子密度以及自旋极化效应。我们发现,每个双自由基异构体的单重态包含两个显着加权的构型,并且较大的活性空间对于正确地表征单重态和三重态都必不可少。2,3-二氢吡嗪的单重态-三重态分裂很小(ortho)和2,6-didehydropyrazine(meta)异构体(分别为+1.8和-1.4 kcal / mol),对于2,5-didehydropyrazine(para)异构体(+28.2 kcal / mol)显着。单重态的几何形状显示了2,3-二氢吡嗪(邻位)和2,6-二氢吡嗪(间位)异构体中的脱氢碳原子之间的全空间相互作用。对有效未成对电子的分析表明,2,5-二氢吡嗪(对)异构体在双自由基电子之间也显示出键间耦合。






























 京公网安备 11010802027423号
京公网安备 11010802027423号