Scientific Reports ( IF 3.8 ) Pub Date : 2019-02-14 , DOI: 10.1038/s41598-019-39391-z Elena A Vidal 1, 2, 3 , Tomás C Moyano 1, 2 , Bernabé I Bustos 1, 4 , Eduardo Pérez-Palma 1, 4 , Carol Moraga 1, 2 , Eleodoro Riveras 1, 5 , Alejandro Montecinos 1, 2 , Lorena Azócar 1, 5 , Daniela C Soto 1, 2 , Mabel Vidal 1, 2 , Alex Di Genova 1, 6 , Klaus Puschel 7 , Peter Nürnberg 8 , Stephan Buch 9 , Jochen Hampe 9 , Miguel L Allende 1, 10 , Verónica Cambiazo 1, 11 , Mauricio González 1, 11 , Christian Hodar 1, 11 , Martín Montecino 1, 4 , Claudia Muñoz-Espinoza 1, 12 , Ariel Orellana 1, 12 , Angélica Reyes-Jara 1, 11 , Dante Travisany 1, 6 , Paula Vizoso 1, 13 , Mauricio Moraga 14, 15 , Susana Eyheramendy 16 , Alejandro Maass 1, 7 , Giancarlo V De Ferrari 1, 4 , Juan Francisco Miquel 1, 5 , Rodrigo A Gutiérrez 1, 2
|
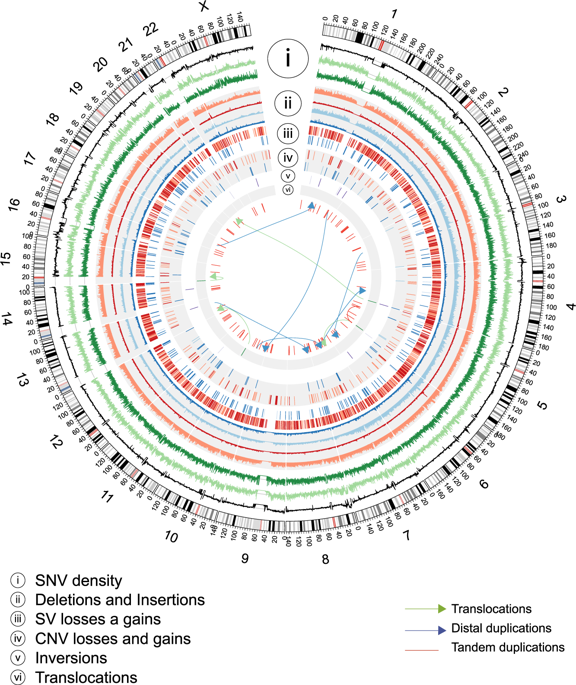
Whole human genome sequencing initiatives help us understand population history and the basis of genetic diseases. Current data mostly focuses on Old World populations, and the information of the genomic structure of Native Americans, especially those from the Southern Cone is scant. Here we present annotation and variant discovery from high-quality complete genome sequences of a cohort of 11 Mapuche-Huilliche individuals (HUI) from Southern Chile. We found approximately 3.1 × 106 single nucleotide variants (SNVs) per individual and identified 403,383 (6.9%) of novel SNVs events. Analyses of large-scale genomic events detected 680 copy number variants (CNVs) and 4,514 structural variants (SVs), including 398 and 1,910 novel events, respectively. Global ancestry composition of HUI genomes revealed that the cohort represents a sample from a marginally admixed population from the Southern Cone, whose main genetic component derives from Native American ancestors. Additionally, we found that HUI genomes contain variants in genes associated with 5 of the 6 leading causes of noncommunicable diseases in Chile, which may have an impact on the risk of prevalent diseases in Chilean and Amerindian populations. Our data represents a useful resource that can contribute to population-based studies and for the design of early diagnostics or prevention tools for Native and admixed Latin American populations.
中文翻译:
南美马普切-韦利切原住民的全基因组序列、变异发现和注释。
全人类基因组测序计划帮助我们了解人口历史和遗传疾病的基础。目前的数据主要集中在旧世界人口,美洲原住民,尤其是来自南锥体的原住民的基因组结构信息很少。在这里,我们展示了来自智利南部 11 名马普切-惠利切个体 (HUI) 的高质量完整基因组序列的注释和变异发现。我们发现每个个体大约有 3.1 × 10 6 个单核苷酸变异 (SNV),并鉴定了 403,383 (6.9%) 个新的 SNV 事件。对大规模基因组事件的分析检测到 680 个拷贝数变异 (CNV) 和 4,514 个结构变异 (SV),分别包括 398 个和 1,910 个新事件。 HUI 基因组的全球血统组成表明,该群体代表了来自南锥体的略微混合人群的样本,其主要遗传成分源自美洲原住民祖先。此外,我们发现 HUI 基因组包含与智利 6 种主要非传染性疾病病因中的 5 种相关的基因变异,这可能对智利和美洲印第安人人群流行疾病的风险产生影响。我们的数据代表了一种有用的资源,可以有助于基于人群的研究以及为拉丁美洲原住民和混合人群设计早期诊断或预防工具。






























 京公网安备 11010802027423号
京公网安备 11010802027423号