当前位置:
X-MOL 学术
›
Free Radical Bio. Med.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
CD36 dependent redoxosomes promotes ceramide-mediated pancreatic β-cell failure via p66Shc activation.
Free Radical Biology and Medicine ( IF 7.1 ) Pub Date : 2019-02-05 , DOI: 10.1016/j.freeradbiomed.2019.02.004
Udayakumar Karunakaran 1 , Suma Elumalai 2 , Jun Sung Moon 1 , Kyu Chang Won 3
Free Radical Biology and Medicine ( IF 7.1 ) Pub Date : 2019-02-05 , DOI: 10.1016/j.freeradbiomed.2019.02.004
Udayakumar Karunakaran 1 , Suma Elumalai 2 , Jun Sung Moon 1 , Kyu Chang Won 3
Affiliation
|
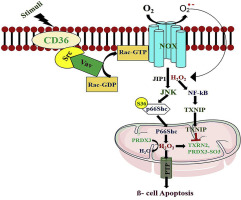
Altered metabolism is implicated in the pathogenesis of beta-cell failure in type 2 diabetes (T2D). Plasma and tissue levels of ceramide species play positive roles in inflammatory and oxidative stress responses in T2D. However, oxidative targets and mechanisms underlying ceramide signaling are unclear. We investigated the role of CD36-dependent redoxosome (redox-active endosome), a membrane-based signaling agent, in ceramide-induced beta-cell dysfunction and failure. Exposure of beta cells to C2-ceramide (N-acetyl-sphingosine) induced a CD36-dependent non-receptor tyrosine kinase Src-mediated redoxosome (Vav2-Rac1-NOX) formation. Activated Rac1-GTP-NADPH oxidase complex induced c-Jun-N-terminal kinase (JNK) activation and nuclear factor (NF)-kB transcription, which was associated with thioredoxin-interacting protein (TXNIP) upregulation and thioredoxin activity suppression. Upregulated JNK expression induced p66Shc serine36 phosphorylation and peroxiredoxin-3 hyperoxidation, causing beta-cell apoptosis via mitochondrial dysfunction. CD36 inhibition by sulfo-N-succinimidyl oleate (SSO) or CD36 siRNA blocked C2-ceramide-induced redoxosome activation, thereby decreasing JNK-dependent p66Shc serine36 phosphorylation. CD36 inhibition downregulated TXNIP expression and promoted thioredoxin activity via enhanced thioredoxin reductase activity, which prevented peroxiredoxin-3 oxidation. CD36 inhibition potentiated glucose-stimulated insulin secretion and prevented beta-cell apoptosis. Our results reveal a new role of CD36 during early molecular events that lead to Src-mediated redoxosome activation, which contributes to ceramide-induced pancreatic beta-cell dysfunction and failure.
中文翻译:

CD36依赖的氧化还原体通过p66Shc激活促进神经酰胺介导的胰腺β细胞衰竭。
新陈代谢的改变与2型糖尿病(T2D)中β细胞衰竭的发病机制有关。在T2D中,神经酰胺种类的血浆和组织水平在炎症和氧化应激反应中发挥积极作用。然而,尚不清楚神经酰胺信号转导的氧化靶点和机制。我们调查了CD36依赖的氧化还原体(氧化还原活性内体),一种基于膜的信号传导剂,在神经酰胺诱导的β细胞功能障碍和衰竭中的作用。β细胞暴露于C2-神经酰胺(N-乙酰基-鞘氨醇)诱导CD36依赖性非受体酪氨酸激酶Src介导的氧化还原体(Vav2-Rac1-NOX)的形成。活化的Rac1-GTP-NADPH氧化酶复合物诱导c-Jun-N-末端激酶(JNK)活化和核因子(NF)-kB转录,这与硫氧还蛋白相互作用蛋白(TXNIP)的上调和硫氧还蛋白的活性抑制有关。JNK表达上调诱导p66Shc丝氨酸36磷酸化和peroxiredoxin-3过氧化,通过线粒体功能障碍导致β细胞凋亡。磺基-N-琥珀酰亚胺基油酸酯(SSO)或CD36 siRNA对CD36的抑制作用阻止了C2-神经酰胺诱导的氧化还原体的活化,从而降低了JNK依赖性p66Shc丝氨酸36的磷酸化。CD36抑制通过增强硫氧还蛋白还原酶活性来下调TXNIP表达并促进硫氧还蛋白活性,从而防止过氧化物酶3的氧化。CD36抑制增强了葡萄糖刺激的胰岛素分泌,并阻止了β细胞凋亡。我们的结果揭示了CD36在早期分子事件中的新作用,该事件导致Src介导的氧化还原体活化,
更新日期:2019-02-05
中文翻译:
CD36依赖的氧化还原体通过p66Shc激活促进神经酰胺介导的胰腺β细胞衰竭。
新陈代谢的改变与2型糖尿病(T2D)中β细胞衰竭的发病机制有关。在T2D中,神经酰胺种类的血浆和组织水平在炎症和氧化应激反应中发挥积极作用。然而,尚不清楚神经酰胺信号转导的氧化靶点和机制。我们调查了CD36依赖的氧化还原体(氧化还原活性内体),一种基于膜的信号传导剂,在神经酰胺诱导的β细胞功能障碍和衰竭中的作用。β细胞暴露于C2-神经酰胺(N-乙酰基-鞘氨醇)诱导CD36依赖性非受体酪氨酸激酶Src介导的氧化还原体(Vav2-Rac1-NOX)的形成。活化的Rac1-GTP-NADPH氧化酶复合物诱导c-Jun-N-末端激酶(JNK)活化和核因子(NF)-kB转录,这与硫氧还蛋白相互作用蛋白(TXNIP)的上调和硫氧还蛋白的活性抑制有关。JNK表达上调诱导p66Shc丝氨酸36磷酸化和peroxiredoxin-3过氧化,通过线粒体功能障碍导致β细胞凋亡。磺基-N-琥珀酰亚胺基油酸酯(SSO)或CD36 siRNA对CD36的抑制作用阻止了C2-神经酰胺诱导的氧化还原体的活化,从而降低了JNK依赖性p66Shc丝氨酸36的磷酸化。CD36抑制通过增强硫氧还蛋白还原酶活性来下调TXNIP表达并促进硫氧还蛋白活性,从而防止过氧化物酶3的氧化。CD36抑制增强了葡萄糖刺激的胰岛素分泌,并阻止了β细胞凋亡。我们的结果揭示了CD36在早期分子事件中的新作用,该事件导致Src介导的氧化还原体活化,
































 京公网安备 11010802027423号
京公网安备 11010802027423号