当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The Coupling between Stability and Ion Pair Formation in Magnesium Electrolytes from First-Principles Quantum Mechanics and Classical Molecular Dynamics
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2015-02-24 , DOI: 10.1021/jacs.5b01004 Nav Nidhi Rajput 1 , Xiaohui Qu 1 , Niya Sa 2 , Anthony K. Burrell 2 , Kristin A. Persson 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2015-02-24 , DOI: 10.1021/jacs.5b01004 Nav Nidhi Rajput 1 , Xiaohui Qu 1 , Niya Sa 2 , Anthony K. Burrell 2 , Kristin A. Persson 1
Affiliation
|
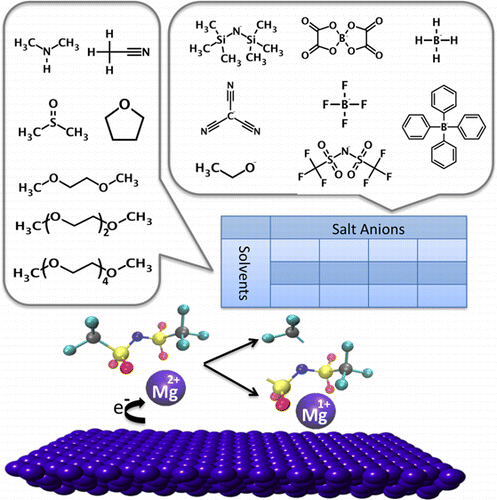
In this work we uncover a novel effect between concentration dependent ion pair formation and anion stability at reducing potentials, e.g., at the metal anode. Through comprehensive calculations using both first-principles as well as well-benchmarked classical molecular dynamics over a matrix of electrolytes, covering solvents and salt anions with a broad range in chemistry, we elucidate systematic correlations between molecular level interactions and composite electrolyte properties, such as electrochemical stability, solvation structure, and dynamics. We find that Mg electrolytes are highly prone to ion pair formation, even at modest concentrations, for a wide range of solvents with different dielectric constants, which have implications for dynamics as well as charge transfer. Specifically, we observe that, at Mg metal potentials, the ion pair undergoes partial reduction at the Mg cation center (Mg(2+) → Mg(+)), which competes with the charge transfer mechanism and can activate the anion to render it susceptible to decomposition. Specifically, TFSI(-) exhibits a significant bond weakening while paired with the transient, partially reduced Mg(+). In contrast, BH4(-) and BF4(-) are shown to be chemically stable in a reduced ion pair configuration. Furthermore, we observe that higher order glymes as well as DMSO improve the solubility of Mg salts, but only the longer glyme chains reduce the dynamics of the ions in solution. This information provides critical design metrics for future electrolytes as it elucidates a close connection between bulk solvation and cathodic stability as well as the dynamics of the salt.
中文翻译:

从第一性原理量子力学和经典分子动力学看镁电解质中稳定性与离子对形成的耦合
在这项工作中,我们揭示了浓度依赖性离子对形成与还原电位(例如金属阳极)下的阴离子稳定性之间的新效应。通过在电解质基质上使用第一性原理和经过良好基准测试的经典分子动力学进行综合计算,涵盖化学范围广泛的溶剂和盐阴离子,我们阐明了分子级相互作用和复合电解质特性之间的系统相关性,例如电化学稳定性、溶剂化结构和动力学。我们发现,对于具有不同介电常数的各种溶剂,Mg 电解质极易形成离子对,即使浓度适中,这对动力学和电荷转移都有影响。具体来说,我们观察到,在 Mg 金属电位下,离子对在 Mg 阳离子中心发生部分还原(Mg(2+) → Mg(+)),这与电荷转移机制竞争并可以激活阴离子使其易于分解。具体而言,TFSI(-) 表现出显着的键弱化,同时与瞬态、部分还原的 Mg(+) 配对。相比之下,BH4(-) 和 BF4(-) 在减少的离子对配置中表现出化学稳定性。此外,我们观察到高阶甘醇二甲醚和 DMSO 提高了镁盐的溶解度,但只有较长的甘醇二甲醚链会降低溶液中离子的动力学。该信息为未来的电解质提供了关键的设计指标,因为它阐明了本体溶剂化和阴极稳定性以及盐的动力学之间的密切联系。
更新日期:2015-02-24
中文翻译:
从第一性原理量子力学和经典分子动力学看镁电解质中稳定性与离子对形成的耦合
在这项工作中,我们揭示了浓度依赖性离子对形成与还原电位(例如金属阳极)下的阴离子稳定性之间的新效应。通过在电解质基质上使用第一性原理和经过良好基准测试的经典分子动力学进行综合计算,涵盖化学范围广泛的溶剂和盐阴离子,我们阐明了分子级相互作用和复合电解质特性之间的系统相关性,例如电化学稳定性、溶剂化结构和动力学。我们发现,对于具有不同介电常数的各种溶剂,Mg 电解质极易形成离子对,即使浓度适中,这对动力学和电荷转移都有影响。具体来说,我们观察到,在 Mg 金属电位下,离子对在 Mg 阳离子中心发生部分还原(Mg(2+) → Mg(+)),这与电荷转移机制竞争并可以激活阴离子使其易于分解。具体而言,TFSI(-) 表现出显着的键弱化,同时与瞬态、部分还原的 Mg(+) 配对。相比之下,BH4(-) 和 BF4(-) 在减少的离子对配置中表现出化学稳定性。此外,我们观察到高阶甘醇二甲醚和 DMSO 提高了镁盐的溶解度,但只有较长的甘醇二甲醚链会降低溶液中离子的动力学。该信息为未来的电解质提供了关键的设计指标,因为它阐明了本体溶剂化和阴极稳定性以及盐的动力学之间的密切联系。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号