Cell Systems ( IF 9.0 ) Pub Date : 2017-09-27 , DOI: 10.1016/j.cels.2017.09.001 Sheng Wang 1 , Zhen Li 2 , Yizhou Yu 3 , Jinbo Xu 4
|
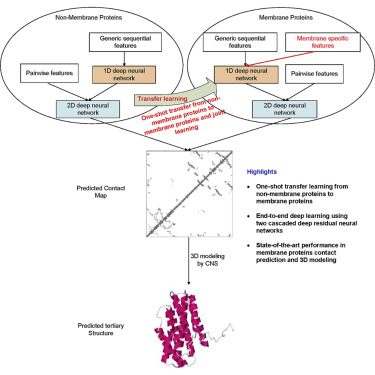
Computational elucidation of membrane protein (MP) structures is challenging partially due to lack of sufficient solved structures for homology modeling. Here, we describe a high-throughput deep transfer learning method that first predicts MP contacts by learning from non-MPs and then predicts 3D structure models using the predicted contacts as distance restraints. Tested on 510 non-redundant MPs, our method has contact prediction accuracy at least 0.18 better than existing methods, predicts correct folds for 218 MPs, and generates 3D models with root-mean-square deviation (RMSD) less than 4 and 5 Å for 57 and 108 MPs, respectively. A rigorous blind test in the continuous automated model evaluation project shows that our method predicted high-resolution 3D models for two recent test MPs of 210 residues with RMSD ∼2 Å. We estimated that our method could predict correct folds for 1,345–1,871 reviewed human multi-pass MPs including a few hundred new folds, which shall facilitate the discovery of drugs targeting at MPs.
中文翻译:
通过深度迁移学习折叠膜蛋白
膜蛋白(MP)结构的计算阐明具有挑战性,部分原因是缺乏足够的同源建模解析结构。在这里,我们描述了一种高通量深度迁移学习方法,该方法首先通过向非 MP 学习来预测 MP 接触,然后使用预测的接触作为距离限制来预测 3D 结构模型。在 510 个非冗余 MP 上进行测试,我们的方法的接触预测精度比现有方法至少提高 0.18,预测 218 个 MP 的正确折叠,并生成均方根偏差 (RMSD) 小于 4 和 5 Å 的 3D 模型分别有 57 名和 108 名议员。连续自动化模型评估项目中的严格盲测表明,我们的方法预测了两个最近的 210 个残基测试 MP 的高分辨率 3D 模型,RMSD ∼2 Å。我们估计我们的方法可以预测 1,345-1,871 个经过审查的人类多次 MP 的正确折叠,其中包括数百个新折叠,这将有助于发现针对 MP 的药物。






























 京公网安备 11010802027423号
京公网安备 11010802027423号