当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
三乙铵三氟甲磺酸质子离子液体的密度泛函计算和分子动力学模拟
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2017-12-11 00:00:00 , DOI: 10.1021/acs.jpcb.7b10373
Juan F. Mora Cardozo 1 , T. Burankova 1 , J. P. Embs 1 , A. Benedetto 1, 2, 3 , P. Ballone 4
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2017-12-11 00:00:00 , DOI: 10.1021/acs.jpcb.7b10373
Juan F. Mora Cardozo 1 , T. Burankova 1 , J. P. Embs 1 , A. Benedetto 1, 2, 3 , P. Ballone 4
Affiliation
|
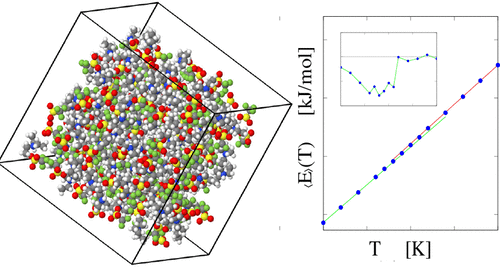
系统的分子动力学模拟基于经验力场已经进行了用于三乙基三氟甲磺酸酯的样品(三乙三氟甲磺酸酯,[TEA] [TF]),覆盖一个宽的温度范围为200K≤ Ť ≤400 K和分析了广泛的一组属性从自扩散和电导率到旋转弛豫和氢键动力学。这项研究是由最近准弹性中子散射和在同一系统上差示扫描量热法测量的动机,揭示在两个连续的一阶转变Ť ≈230和310 K(加热),以及一个有趣的和部分不明原因的各种subdiffusive的酸性质子的运动。仿真显示在T处有一个弱的不连续过渡= 310 K,并突出显示了离子的旋转弛豫中T = 260 K处的异常,这是我们在T = 230 K处的实验跃迁的模拟模拟中识别出的。因此,模拟有助于识别实验跃迁的性质,从而确认了最高温度之一对应于熔化,而在较低的一个正在发生Ť是从晶体的过渡,稳定在Ť ≤260 K,在塑料相(260≤ Ť ≤310 K),其中分子能够旋转而不扩散。尤其是旋转导致了中间T处的亚扩散运动无论是在实验中还是在仿真中。通过分子动力学和密度泛函计算研究了氢键的结构,分布和强度。还讨论了具有相同符号的离子的聚集以及水在1%wgt浓度下被水污染的影响。

"点击查看英文标题和摘要"
更新日期:2017-12-11
"点击查看英文标题和摘要"



































 京公网安备 11010802027423号
京公网安备 11010802027423号