当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Universal Dynamics of Molecular Reorientation in Hybrid Lead Iodide Perovskites
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2017-11-02 , DOI: 10.1021/jacs.7b09536
Douglas H. Fabini 1 , Ting Ann Siaw 2 , Constantinos C. Stoumpos 3 , Geneva Laurita 4 , Daniel Olds 5 , Katharine Page 5 , Jerry G. Hu 1 , Mercouri G. Kanatzidis 3 , Songi Han 2, 6 , Ram Seshadri 1, 2
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2017-11-02 , DOI: 10.1021/jacs.7b09536
Douglas H. Fabini 1 , Ting Ann Siaw 2 , Constantinos C. Stoumpos 3 , Geneva Laurita 4 , Daniel Olds 5 , Katharine Page 5 , Jerry G. Hu 1 , Mercouri G. Kanatzidis 3 , Songi Han 2, 6 , Ram Seshadri 1, 2
Affiliation
|
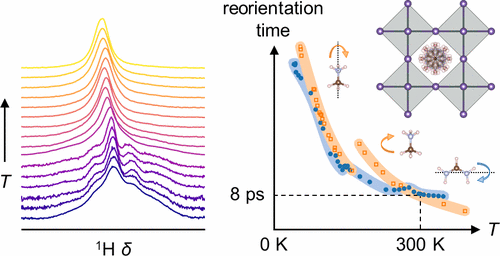
The role of organic molecular cations in the high-performance perovskite photovoltaic absorbers, methylammonium lead iodide (MAPbI3) and formamidinium lead iodide (FAPbI3), has been an enigmatic subject of great interest. Beyond aiding in the ease of processing of thin films for photovoltaic devices, there have been suggestions that many of the remarkable properties of the halide perovskites can be attributed to the dipolar nature and the dynamic behavior of these cations. Here, we establish the dynamics of the molecular cations in FAPbI3 between 4 K and 340 K and the nature of their interaction with the surrounding inorganic cage using a combination of solid state nuclear magnetic resonance and dielectric spectroscopies, neutron scattering, calorimetry, and ab initio calculations. Detailed comparisons with the reported temperature dependence of the dynamics of MAPbI3 are then carried out which reveal the molecular ions in the two different compounds to exhibit very similar rotation rates (≈8 ps) at room temperature, despite differences in other temperature regimes. For FA, rotation about the N···N axis, which reorients the molecular dipole, is the dominant motion in all phases, with an activation barrier of ≈21 meV in the ambient phase, compared to ≈110 meV for the analogous dipole reorientation of MA. Geometrical frustration of the molecule-cage interaction in FAPbI3 produces a disordered γ-phase and subsequent glassy freezing at yet lower temperatures. Hydrogen bonds suggested by atom-atom distances from neutron total scattering experiments imply a substantial role for the molecules in directing structure and dictating properties. The temperature dependence of reorientation of the dipolar molecular cations systematically described here can clarify various hypotheses including those of large-polaron charge transport and fugitive electron spin polarization that have been invoked in the context of these unusual materials.
中文翻译:

混合碘化铅钙钛矿中分子重新定向的通用动力学
有机分子阳离子在高性能钙钛矿光伏吸收剂、甲基铵碘化铅 (MAPbI3) 和甲脒碘化铅 (FAPbI3) 中的作用一直是一个令人非常感兴趣的神秘主题。除了有助于简化光伏器件薄膜的加工外,有人认为卤化物钙钛矿的许多显着特性可归因于这些阳离子的偶极性质和动态行为。在这里,我们使用固态核磁共振和介电光谱、中子散射、量热法和 ab initio 的组合,建立了 4 K 和 340 K 之间 FAPbI3 中分子阳离子的动力学及其与周围无机笼相互作用的性质。计算。然后进行与报道的 MAPbI3 动力学的温度依赖性的详细比较,这揭示了两种不同化合物中的分子离子在室温下表现出非常相似的旋转速率(≈8 ps),尽管其他温度范围存在差异。对于 FA,围绕 N…N 轴的旋转使分子偶极子重新定向,是所有相中的主要运动,环境相中的激活势垒为 ≈21 meV,而类似的偶极子重新定向为 ≈110 meV马。FAPbI3 中分子-笼相互作用的几何受挫会产生无序的 γ 相和随后在较低温度下的玻璃态冻结。来自中子总散射实验的原子-原子距离所暗示的氢键意味着分子在指导结构和决定性质方面的重要作用。这里系统地描述的偶极分子阳离子重新定向的温度依赖性可以澄清各种假设,包括在这些不寻常材料的背景下调用的大极化子电荷传输和逃逸电子自旋极化的假设。
更新日期:2017-11-02
中文翻译:
混合碘化铅钙钛矿中分子重新定向的通用动力学
有机分子阳离子在高性能钙钛矿光伏吸收剂、甲基铵碘化铅 (MAPbI3) 和甲脒碘化铅 (FAPbI3) 中的作用一直是一个令人非常感兴趣的神秘主题。除了有助于简化光伏器件薄膜的加工外,有人认为卤化物钙钛矿的许多显着特性可归因于这些阳离子的偶极性质和动态行为。在这里,我们使用固态核磁共振和介电光谱、中子散射、量热法和 ab initio 的组合,建立了 4 K 和 340 K 之间 FAPbI3 中分子阳离子的动力学及其与周围无机笼相互作用的性质。计算。然后进行与报道的 MAPbI3 动力学的温度依赖性的详细比较,这揭示了两种不同化合物中的分子离子在室温下表现出非常相似的旋转速率(≈8 ps),尽管其他温度范围存在差异。对于 FA,围绕 N…N 轴的旋转使分子偶极子重新定向,是所有相中的主要运动,环境相中的激活势垒为 ≈21 meV,而类似的偶极子重新定向为 ≈110 meV马。FAPbI3 中分子-笼相互作用的几何受挫会产生无序的 γ 相和随后在较低温度下的玻璃态冻结。来自中子总散射实验的原子-原子距离所暗示的氢键意味着分子在指导结构和决定性质方面的重要作用。这里系统地描述的偶极分子阳离子重新定向的温度依赖性可以澄清各种假设,包括在这些不寻常材料的背景下调用的大极化子电荷传输和逃逸电子自旋极化的假设。
































 京公网安备 11010802027423号
京公网安备 11010802027423号