当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Merging Metadynamics into Hyperdynamics: Accelerated Molecular Simulations Reaching Time Scales from Microseconds to Seconds
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2015-09-15 00:00:00 , DOI: 10.1021/acs.jctc.5b00597 Kristof M. Bal 1 , Erik C. Neyts 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2015-09-15 00:00:00 , DOI: 10.1021/acs.jctc.5b00597 Kristof M. Bal 1 , Erik C. Neyts 1
Affiliation
|
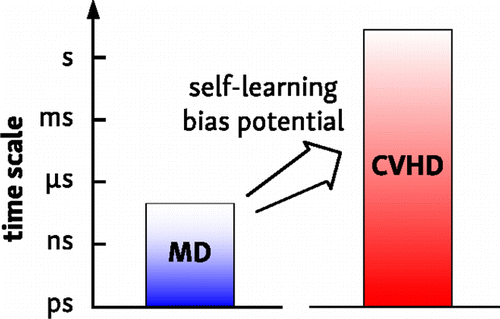
The hyperdynamics method is a powerful tool to simulate slow processes at the atomic level. However, the construction of an optimal hyperdynamics potential is a task that is far from trivial. Here, we propose a generally applicable implementation of the hyperdynamics algorithm, borrowing two concepts from metadynamics. First, the use of a collective variable (CV) to represent the accelerated dynamics gives the method a very large flexibility and simplicity. Second, a metadynamics procedure can be used to construct a suitable history-dependent bias potential on-the-fly, effectively turning the algorithm into a self-learning accelerated molecular dynamics method. This collective variable-driven hyperdynamics (CVHD) method has a modular design: both the local system properties on which the bias is based, as well as the characteristics of the biasing method itself, can be chosen to match the needs of the considered system. As a result, system-specific details are abstracted from the biasing algorithm itself, making it extremely versatile and transparent. The method is tested on three model systems: diffusion on the Cu(001) surface and nickel-catalyzed methane decomposition, as examples of “reactive” processes with a bond-length-based CV, and the folding of a long polymer-like chain, using a set of dihedral angles as a CV. Boost factors up to 109, corresponding to a time scale of seconds, could be obtained while still accurately reproducing correct dynamics.
中文翻译:

将元动力学合并为超动力学:加速分子模拟,时间范围从微秒到秒
超动力学方法是一种功能强大的工具,可以在原子水平上模拟慢速过程。但是,构建最佳的超动力学潜能绝非易事。在这里,我们从超动力学中借鉴了两个概念,提出了超动力学算法的一种普遍适用的实现方式。首先,使用集体变量(CV)表示加速的动力学特性使该方法具有很大的灵活性和简便性。其次,可以使用元动力学过程来动态构建合适的历史相关偏电势,从而有效地将算法转变为自学习的加速分子动力学方法。这种集体变量驱动的超动力学(CVHD)方法采用模块化设计:偏差基于的本地系统属性,以及偏置方法本身的特性,都可以选择以匹配所考虑系统的需求。结果,系统特定的细节从偏差算法本身中抽象出来,使其具有极大的通用性和透明性。该方法在三种模型系统上进行了测试:在Cu(001)表面扩散和镍催化的甲烷分解(作为具有键长的CV的“反应性”过程的示例)以及长聚合物状链的折叠,使用一组二面角作为CV。提升系数高达10 扩散在Cu(001)表面和镍催化的甲烷分解,作为使用基于键长的CV的“反应”过程的示例,以及使用一组二面角作为折叠的长聚合物状链的折叠简历。提升系数高达10 扩散在Cu(001)表面和镍催化的甲烷分解,作为使用基于键长的CV的“反应”过程的示例,以及使用一组二面角作为折叠的长聚合物状链的折叠简历。提升系数高达10在仍然准确地再现正确的动态的同时,可以获得对应于秒的时间标度的图9。
更新日期:2015-09-15
中文翻译:
将元动力学合并为超动力学:加速分子模拟,时间范围从微秒到秒
超动力学方法是一种功能强大的工具,可以在原子水平上模拟慢速过程。但是,构建最佳的超动力学潜能绝非易事。在这里,我们从超动力学中借鉴了两个概念,提出了超动力学算法的一种普遍适用的实现方式。首先,使用集体变量(CV)表示加速的动力学特性使该方法具有很大的灵活性和简便性。其次,可以使用元动力学过程来动态构建合适的历史相关偏电势,从而有效地将算法转变为自学习的加速分子动力学方法。这种集体变量驱动的超动力学(CVHD)方法采用模块化设计:偏差基于的本地系统属性,以及偏置方法本身的特性,都可以选择以匹配所考虑系统的需求。结果,系统特定的细节从偏差算法本身中抽象出来,使其具有极大的通用性和透明性。该方法在三种模型系统上进行了测试:在Cu(001)表面扩散和镍催化的甲烷分解(作为具有键长的CV的“反应性”过程的示例)以及长聚合物状链的折叠,使用一组二面角作为CV。提升系数高达10 扩散在Cu(001)表面和镍催化的甲烷分解,作为使用基于键长的CV的“反应”过程的示例,以及使用一组二面角作为折叠的长聚合物状链的折叠简历。提升系数高达10 扩散在Cu(001)表面和镍催化的甲烷分解,作为使用基于键长的CV的“反应”过程的示例,以及使用一组二面角作为折叠的长聚合物状链的折叠简历。提升系数高达10在仍然准确地再现正确的动态的同时,可以获得对应于秒的时间标度的图9。










































 京公网安备 11010802027423号
京公网安备 11010802027423号