当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Unraveling the Mechanism of the Initiation Reaction of the Methanol to Olefins Process Using ab Initio and DFT Calculations
ACS Catalysis ( IF 11.3 ) Pub Date : 2017-10-26 00:00:00 , DOI: 10.1021/acscatal.7b03114 Philipp N. Plessow 1 , Felix Studt 1, 2
ACS Catalysis ( IF 11.3 ) Pub Date : 2017-10-26 00:00:00 , DOI: 10.1021/acscatal.7b03114 Philipp N. Plessow 1 , Felix Studt 1, 2
Affiliation
|
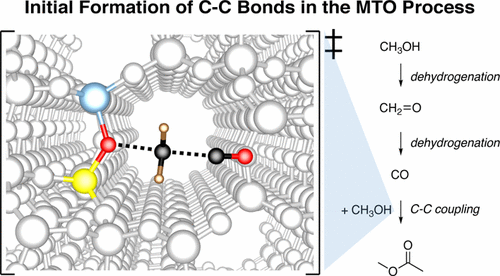
We report a theoretical investigation of the initiation of the methanol to olefin process, where we study the full reaction mechanism from methanol to propylene. The zeolite H-SSZ-13 is investigated with periodic density functional theory (DFT) calculations. These calculations are corrected with MP2-calculations on large (46T) cluster models, which is found to be crucial for sufficient accuracy. Our calculations clearly demonstrate that initiation via the formation of carbon monoxide is a realistic mechanism and is more likely than the methane–formaldehyde mechanism or variants thereof. A kinetic model of the autocatalytic carbon pool mechanism is employed to investigate the initiation kinetics in more detail, demonstrating that an assessment of the feasibility of an initiation reaction needs to be based on kinetic modeling of both the initiation reaction and autocatalysis. This model gives further evidence that initiation proceeds via oxidation of methanol to carbon monoxide, which subsequently forms the first carbon–carbon bond via carbonylation of methanol. The kinetic model also shows that only extremely small amounts of an olefin need to be formed for autocatalysis to start, implying that small impurities will dominate over initiation mechanisms.
中文翻译:

使用从头算和DFT计算揭示甲醇引发烯烃反应的机理
我们报告了从甲醇到烯烃的引发过程的理论研究,在这里我们研究了从甲醇到丙烯的完整反应机理。使用周期性密度泛函理论(DFT)计算研究了H-SSZ-13沸石。这些计算已通过大型(46T)群集模型上的MP2计算进行了修正,这对于确保足够的准确性至关重要。我们的计算清楚地表明,通过一氧化碳的形成引发是一个现实的机制,并且比甲烷-甲醛机制或其变体更有可能。使用自动催化碳库机制的动力学模型来更详细地研究引发动力学,证明对引发反应可行性的评估需要基于引发反应和自催化的动力学建模。该模型提供了进一步的证据,表明引发是通过甲醇氧化为一氧化碳而进行的,而一氧化碳随后通过甲醇的羰基化反应形成第一个碳-碳键。动力学模型还表明,仅需要形成极少量的烯烃即可开始自动催化,这意味着小杂质将在引发机制中占主导地位。
更新日期:2017-10-26
中文翻译:
使用从头算和DFT计算揭示甲醇引发烯烃反应的机理
我们报告了从甲醇到烯烃的引发过程的理论研究,在这里我们研究了从甲醇到丙烯的完整反应机理。使用周期性密度泛函理论(DFT)计算研究了H-SSZ-13沸石。这些计算已通过大型(46T)群集模型上的MP2计算进行了修正,这对于确保足够的准确性至关重要。我们的计算清楚地表明,通过一氧化碳的形成引发是一个现实的机制,并且比甲烷-甲醛机制或其变体更有可能。使用自动催化碳库机制的动力学模型来更详细地研究引发动力学,证明对引发反应可行性的评估需要基于引发反应和自催化的动力学建模。该模型提供了进一步的证据,表明引发是通过甲醇氧化为一氧化碳而进行的,而一氧化碳随后通过甲醇的羰基化反应形成第一个碳-碳键。动力学模型还表明,仅需要形成极少量的烯烃即可开始自动催化,这意味着小杂质将在引发机制中占主导地位。




























 京公网安备 11010802027423号
京公网安备 11010802027423号