当前位置:
X-MOL 学术
›
J. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
On sulfur core level binding energies in thiol self-assembly and alternative adsorption sites: An experimental and theoretical study
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2015-09-10 13:05:13 , DOI: 10.1063/1.4929350
Juanjuan Jia 1, 2 , Abdelkader Kara 3 , Luca Pasquali 4, 5, 6 , Azzedine Bendounan 7 , Fausto Sirotti 7 , Vladimir A. Esaulov 1, 2, 5
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2015-09-10 13:05:13 , DOI: 10.1063/1.4929350
Juanjuan Jia 1, 2 , Abdelkader Kara 3 , Luca Pasquali 4, 5, 6 , Azzedine Bendounan 7 , Fausto Sirotti 7 , Vladimir A. Esaulov 1, 2, 5
Affiliation
|
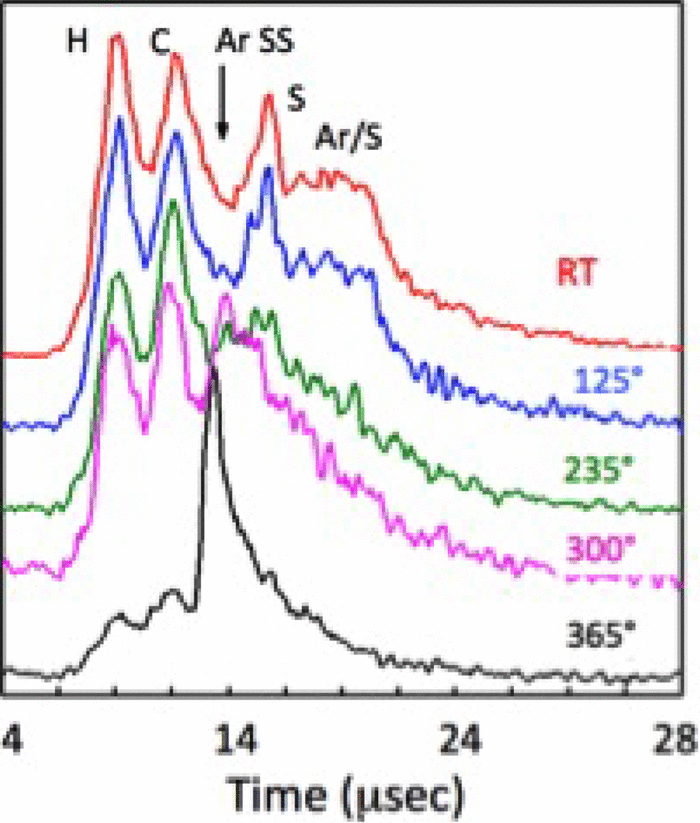
Characteristic core level binding energies (CLBEs) are regularly used to infer the modes of molecular adsorption: orientation, organization, and dissociation processes. Here, we focus on a largely debated situation regarding CLBEs in the case of chalcogen atom bearing molecules. For a thiol, this concerns the case when the CLBE of a thiolate sulfur at an adsorption site can be interpreted alternatively as due to atomic adsorption of a S atom, resulting from dissociation. Results of an investigation of the characteristics of thiol self-assembled monolayers (SAMs) obtained by vacuum evaporative adsorption are presented along with core level binding energy calculations. Thiol ended SAMs of 1,4-benzenedimethanethiol (BDMT) obtained by evaporation on Au display an unconventional CLBE structure at about 161.25 eV, which is close to a known CLBE of a S atom on Au. Adsorption and CLBE calculations for sulfur atoms and BDMT molecules are reported and allow delineating trends as a function of chemisorption on hollow, bridge, and atop sites and including the presence of adatoms. These calculations suggest that the 161.25 eV peak is due to an alternative adsorption site, which could be associated to an atop configuration. Therefore, this may be an alternative interpretation, different from the one involving the adsorption of atomic sulfur resulting from the dissociation process of the S–C bond. Calculated differences in S(2p) CLBEs for free BDMT molecules, SH group sulfur on top of the SAM, and disulfide are also reported to clarify possible errors in assignments.
中文翻译:

硫醇自组装和替代吸附位中硫核能级结合能的实验和理论研究
经常使用特征性的核心能级结合能(CLBE)来推断分子吸附的模式:取向,组织和解离过程。在这里,我们集中讨论在含硫族元素原子的分子中关于CLBE的广泛争议的情况。对于硫醇,这涉及以下情况:硫醇盐硫在吸附位点的CLBE可以替代地解释为由于解离产生的S原子的原子吸附。提出了对通过真空蒸发吸附获得的硫醇自组装单分子膜(SAMs)的特性进行研究的结果,并给出了核心能级结合能的计算结果。通过在Au上蒸发获得的1,4-苯二甲硫醇(BDMT)的硫醇末端SAM在约161.25 eV处显示出非常规的CLBE结构,它接近于Au上S原子的已知CLBE。已报道了硫原子和BDMT分子的吸附和CLBE计算,并可以根据中空,桥和顶部的化学吸附(包括吸附原子的存在)来描绘趋势。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。已报道了硫原子和BDMT分子的吸附和CLBE计算,并可以根据中空,桥和顶部的化学吸附(包括吸附原子的存在)来描绘趋势。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。已报道了硫原子和BDMT分子的吸附和CLBE计算,并可以根据中空,桥和顶部的化学吸附(包括吸附原子的存在)来描绘趋势。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。
更新日期:2015-09-11
中文翻译:
硫醇自组装和替代吸附位中硫核能级结合能的实验和理论研究
经常使用特征性的核心能级结合能(CLBE)来推断分子吸附的模式:取向,组织和解离过程。在这里,我们集中讨论在含硫族元素原子的分子中关于CLBE的广泛争议的情况。对于硫醇,这涉及以下情况:硫醇盐硫在吸附位点的CLBE可以替代地解释为由于解离产生的S原子的原子吸附。提出了对通过真空蒸发吸附获得的硫醇自组装单分子膜(SAMs)的特性进行研究的结果,并给出了核心能级结合能的计算结果。通过在Au上蒸发获得的1,4-苯二甲硫醇(BDMT)的硫醇末端SAM在约161.25 eV处显示出非常规的CLBE结构,它接近于Au上S原子的已知CLBE。已报道了硫原子和BDMT分子的吸附和CLBE计算,并可以根据中空,桥和顶部的化学吸附(包括吸附原子的存在)来描绘趋势。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。已报道了硫原子和BDMT分子的吸附和CLBE计算,并可以根据中空,桥和顶部的化学吸附(包括吸附原子的存在)来描绘趋势。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。已报道了硫原子和BDMT分子的吸附和CLBE计算,并可以根据中空,桥和顶部的化学吸附(包括吸附原子的存在)来描绘趋势。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。这些计算表明,161.25 eV峰是由于另一个吸附位点所致,该位置可能与顶部构型有关。因此,这可能是另一种解释,不同于涉及由S–C键解离过程导致的原子硫吸附的解释。还报告了针对游离BDMT分子,SAM顶部的SH基硫和二硫键的S(2p)CLBE的计算差异,以阐明分配中的可能错误。
































 京公网安备 11010802027423号
京公网安备 11010802027423号