European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2017-09-21 , DOI: 10.1016/j.ejmech.2017.09.038
Ying-Chao Duan , Yong-Cheng Ma , Wen-Ping Qin , Li-Na Ding , Yi-Chao Zheng , Ying-Li Zhu , Xiao-Yu Zhai , Jing Yang , Chao-Ya Ma , Yuan-Yuan Guan
|
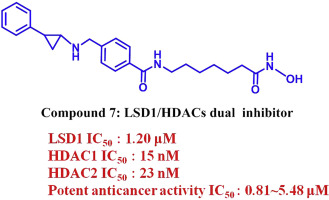
Lysine specific demethylase 1 (LSD1) and Histone deacetylases (HDACs) are promising drug targets for cancers. Recent studies reveal an important functional interplay between LSD1 and HDACs, and there is evidence for the synergistic effect of combined LSD1 and HDAC inhibitors on cancers. Therefore, development of inhibitors targeting both LSD1 and HDACs might be a promising strategy for epigenetic therapy of cancers. We report herein the synthesis of a series of tranylcypromine derivatives as LSD1/HDACs dual inhibitors. Most compounds showed potent LSD1 and HDACs inhibitory activity, especially compound 7 displayed the most potent inhibitory activity against HDAC1 and HDAC2 with IC50 of 15 nM and 23 nM, as well as potent inhibition against LSD1 with IC50 of 1.20 μM. Compound 7 demonstrated stronger anti-proliferative activities than SAHA with IC50 values ranging from 0.81 to 4.28 μM against MGC-803, MCF-7, SW-620 and A-549 human cancer cell lines. Further mechanistic studies showed that compound 7 treatment in MGC-803 cells dose-dependently increased cellular H3K4 and H3K9 methylation, as well as H3 acetylation, decreased the mitochondrial membrane potential and induced remarkable apoptosis. Docking studies showed that compound 7 can be well docked into the active binding sites of LSD1 and HDAC2. This finding highlights the potential for the development of LSD1/HDACs dual inhibitors as novel anticancer drugs.
中文翻译:
设计和合成作为新型LSD1 / HDACs双重抑制剂的tranylcypromine衍生物,用于癌症治疗
赖氨酸特异性脱甲基酶1(LSD1)和组蛋白脱乙酰基酶(HDACs)是有望成为癌症的药物靶标。最近的研究揭示了LSD1和HDAC之间的重要功能相互作用,并且有证据表明LSD1和HDAC组合抑制剂对癌症具有协同作用。因此,针对LSD1和HDAC的抑制剂的开发可能是癌症表观遗传治疗的有前途的策略。我们在此报告了一系列作为LSD1 / HDACs双重抑制剂的反式环丙胺衍生物的合成。大多数化合物显示出对LSD1和HDAC的有效抑制活性,尤其是化合物7对HDAC1和HDAC2表现出最有效的抑制活性,IC 50为15 nM和23 nM,对LSD1的潜在抑制为IC 50为1.20μM。化合物7表现出比SAHA更强的抗增殖活性,针对MGC-803,MCF-7,SW-620和A-549人癌细胞系的IC 50值为0.81至4.28μM。进一步的机理研究表明,化合物7在MGC-803细胞中的处理剂量依赖性地增加了细胞H3K4和H3K9甲基化以及H3乙酰化,从而降低了线粒体膜电位并诱导了显着的细胞凋亡。对接研究表明,化合物7可以很好地对接至LSD1和HDAC2的活性结合位点。这一发现凸显了开发LSD1 / HDACs双重抑制剂作为新型抗癌药物的潜力。
































 京公网安备 11010802027423号
京公网安备 11010802027423号