当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Reaction Paths and Chemical Activation Reactions of 2-Methyl-5-Furanyl Radical with 3O2
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-09-26 00:00:00 , DOI: 10.1021/acs.jpca.7b06650 Jason M. Hudzik 1 , Joseph W. Bozzelli 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-09-26 00:00:00 , DOI: 10.1021/acs.jpca.7b06650 Jason M. Hudzik 1 , Joseph W. Bozzelli 1
Affiliation
|
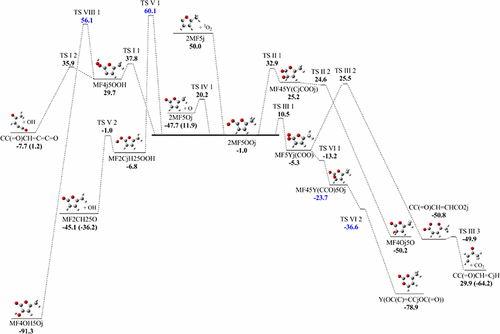
Interest in high-energy substituted furans has been increasing due to their occurrence in biofuel production and their versatility in conversion to other useful products. Methylfurans are the simplest substituted furans and understanding their reaction pathways, thermochemical properties, including intermediate species stability, and chemical kinetics would aid in the study of larger furans. Furan ring C–H bonds have been shown to be extremely strong, approximately 120 kcal mol–1, due in part to the placement of the oxygen atom and aromatic-like resonance, both within the ring. The thermochemistry and kinetics of the oxidation of 2-methyfuran radical at position 5 of the furan ring, 2-methyl-5-furanyl radical (2MF5j), is analyzed. The resulting chemically activated species, 2MF5OOj radical, has a well depth of 51 kcal mol–1 below the 2MF5j + O2 reactants; this is 4–5 kcal mol–1 deeper than that of phenyl and vinyl radical plus O2, with both of these reactions known to undergo chain branching. Important, low-energy reaction pathways include chain branching dissociations, intramolecular abstractions, group transfers, and radical oxygen additions. Enthalpies of formation, entropies, and heat capacities for the stable molecules, radicals, and transition-state species are analyzed using computational methods. Calculated ΔH°f 298 values were determined using an isodesmic work reaction from the CBS-QB3 composite method. Elementary rate parameters are from saddle point transition-state structures and compared to variational transition-state analysis for the barrierless reactions. Temperature- and pressure-dependent rate constants which are calculated using QRRK and master equation analysis is used for falloff and stabilization.
中文翻译:

2-甲基-5-呋喃基自由基与3 O 2的反应路径及化学活化反应
对高能取代呋喃的兴趣由于其在生物燃料生产中的存在及其转化为其他有用产品的多功能性而日益受到关注。甲基呋喃是最简单的取代呋喃,了解它们的反应途径,热化学性质(包括中间物种稳定性)和化学动力学将有助于研究较大的呋喃。呋喃环C–H键已显示出非常强的强度,大约120 kcal mol –1,部分是由于在环内都存在氧原子和类似芳香族的共振。分析了呋喃环第5位的2-甲基呋喃基团2-甲基-5-呋喃基(2MF5j)的氧化反应的热化学和动力学。所得的化学活化物质2MF500j自由基的深度低于2MF5j + O 2反应物的深度为51 kcal mol –1。这比苯基和乙烯基自由基加O 2的深4–5 kcal mol –1,这两个反应都已知会发生链支化。重要的低能反应途径包括链分支解离,分子内抽象,基团转移和自由基氧的添加。使用计算方法分析了稳定分子,自由基和过渡态物种的形成焓,熵和热容。计算的ΔH ° f 298使用来自CBS-QB3复合方法的等电功反应确定值。基本速率参数来自鞍点过渡态结构,并与无障碍反应的变化过渡态分析进行了比较。使用QRRK和主方程分析计算的温度和压力相关的速率常数用于衰减和稳定化。
更新日期:2017-09-26
中文翻译:
2-甲基-5-呋喃基自由基与3 O 2的反应路径及化学活化反应
对高能取代呋喃的兴趣由于其在生物燃料生产中的存在及其转化为其他有用产品的多功能性而日益受到关注。甲基呋喃是最简单的取代呋喃,了解它们的反应途径,热化学性质(包括中间物种稳定性)和化学动力学将有助于研究较大的呋喃。呋喃环C–H键已显示出非常强的强度,大约120 kcal mol –1,部分是由于在环内都存在氧原子和类似芳香族的共振。分析了呋喃环第5位的2-甲基呋喃基团2-甲基-5-呋喃基(2MF5j)的氧化反应的热化学和动力学。所得的化学活化物质2MF500j自由基的深度低于2MF5j + O 2反应物的深度为51 kcal mol –1。这比苯基和乙烯基自由基加O 2的深4–5 kcal mol –1,这两个反应都已知会发生链支化。重要的低能反应途径包括链分支解离,分子内抽象,基团转移和自由基氧的添加。使用计算方法分析了稳定分子,自由基和过渡态物种的形成焓,熵和热容。计算的ΔH ° f 298使用来自CBS-QB3复合方法的等电功反应确定值。基本速率参数来自鞍点过渡态结构,并与无障碍反应的变化过渡态分析进行了比较。使用QRRK和主方程分析计算的温度和压力相关的速率常数用于衰减和稳定化。






























 京公网安备 11010802027423号
京公网安备 11010802027423号