当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Linker Design for Highly Crystalline Metal–Organic Framework NU-1000
Chemistry of Materials ( IF 7.2 ) Pub Date : 2017-09-20 00:00:00 , DOI: 10.1021/acs.chemmater.7b01624 Wei-Guang Liu 1 , Donald G. Truhlar 1
Chemistry of Materials ( IF 7.2 ) Pub Date : 2017-09-20 00:00:00 , DOI: 10.1021/acs.chemmater.7b01624 Wei-Guang Liu 1 , Donald G. Truhlar 1
Affiliation
|
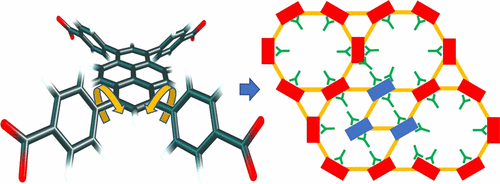
NU-1000 is a metal–organic framework (MOF) with Zr6(μ3–OH)4(μ3–O)4(OH)4(OH2)4 nodes (called Zr6 nodes) and tetratopic 1,3,6,8-tetrakis(p-benzoate)pyrene (TBAPy) linkers. NU-1000 has pores of 31 Å diameter, and it has smaller pores connected perpendicularly to these large pores; this nanoporous structure makes it suitable as a catalyst or a support for catalysis. However, as ordinarily synthesized, NU-1000 has disordered Zr6 nodes populated in its large pores, and this introduces nonuniformity into the crystal and complicates structural characterization. We have examined possible MOF morphologies composed of Zr6 nodes and TBAPy linkers, and we propose that the extra Zr6 nodes in the large pores of NU-1000 are structurally similar to those of an NU-901 phase, which differs from NU-1000 in the relative orientation of Zr6 nodes and the linker conformations. This polymorphism comes from two rotamers of the TBAPy linkers; in NU-1000 the benzoates at the 1 and 3 positions and the 6 and 8 positions are disrotatorily situated, whereas in NU-901 these benzoates are conrotatorily situated. Substituting carbon-2 and carbon-7 of pyrene with groups that are unevenly disposed with respect to the pyrene plane should stabilize the rotamer that can form NU-1000 and therefore stabilize NU-1000 itself. We confirmed this by applying the PBE-D2 and M06-L exchange-correlation functionals in periodic electronic structure calculations to compare the energies of NU-1000 and NU-901 with different substituents on the linkers. In NU-1000, the benzene rings of the benzoates coordinating to the Zr6 nodes form pocketlike structures. In unsubstituted NU-1000 the pocket in the small pore is smaller and attracts precursors of the atomic layer deposition (ALD) step more strongly because of shorter benzene–precursor distances and more favorable dispersion-like interactions, leading to selective metal deposition in the small pores. We examined the pocket sizes and the adsorption energy of Zn(Et)2 (the ALD precursor) on the Zr6 node in large and small pores of substituted NU-1000, and found that bulkier substituents not only compress the pocket in the large pore, but also have shorter distances to the ALD precursor in the large pore. Both factors contribute to the more favorable dispersion interaction and lead to less tendency to deposit metals in the small pore.
中文翻译:

高度结晶的金属有机框架NU-1000的计算链接器设计
NU-1000是一个金属-有机构架(MOF)与Zr的6(μ 3 -OH)4(μ 3 -O)4(OH)4(OH 2)4个节点(称为锆6个节点)和tetratopic 1,3- ,6,8-四(对苯甲酸酯)py(TBAPy)接头。NU-1000的孔直径为31Å,并且垂直于这些大孔的孔较小。这种纳米孔结构使其适合用作催化剂或催化载体。然而,正如通常所合成的,NU-1000的Zr 6无序节点在其大孔中聚集,这将不均匀性引入晶体,并使结构表征复杂化。我们研究了由Zr 6节点和TBAPy接头组成的可能的MOF形态,并且我们认为NU-1000大孔中多余的Zr 6节点在结构上与NU-901相的结构相似,这与NU-1000不同在Zr 6的相对方向节点和链接器构象。这种多态性来自TBAPy接头的两个旋转异构体。在NU-1000中,位于1和3位以及6和8位的苯甲酸酯是旋转位置,而在NU-901中,这些苯甲酸酯是旋转位置。用相对于the平面分布不均的基团取代pyr的碳2和碳7可以稳定可以形成NU-1000的旋转异构体,因此可以稳定NU-1000本身。我们通过在周期性电子结构计算中应用PBE-D2和M06-L交换相关函数来比较连接体上不同取代基的NU-1000和NU-901的能量,从而证实了这一点。在NU-1000中,与Zr 6配位的苯甲酸酯的苯环节点形成袋状结构。在未取代的NU-1000中,小孔中的口袋较小,并且由于较短的苯-前体距离和更有利的类似分散的相互作用,因此更强地吸引了原子层沉积(ALD)步骤的前驱物,从而导致了小金属中的选择性金属沉积。毛孔。我们研究了取代的NU-1000大孔和小孔中Zr 6节点上Zn(Et)2(ALD前体)的袋大小和吸附能,发现更大的取代基不仅压缩了大孔中的袋,但在大孔中距ALD前体的距离也较短。这两个因素有助于更有利的分散相互作用,并导致较小的金属沉积在小孔中的趋势。
更新日期:2017-09-20
中文翻译:
高度结晶的金属有机框架NU-1000的计算链接器设计
NU-1000是一个金属-有机构架(MOF)与Zr的6(μ 3 -OH)4(μ 3 -O)4(OH)4(OH 2)4个节点(称为锆6个节点)和tetratopic 1,3- ,6,8-四(对苯甲酸酯)py(TBAPy)接头。NU-1000的孔直径为31Å,并且垂直于这些大孔的孔较小。这种纳米孔结构使其适合用作催化剂或催化载体。然而,正如通常所合成的,NU-1000的Zr 6无序节点在其大孔中聚集,这将不均匀性引入晶体,并使结构表征复杂化。我们研究了由Zr 6节点和TBAPy接头组成的可能的MOF形态,并且我们认为NU-1000大孔中多余的Zr 6节点在结构上与NU-901相的结构相似,这与NU-1000不同在Zr 6的相对方向节点和链接器构象。这种多态性来自TBAPy接头的两个旋转异构体。在NU-1000中,位于1和3位以及6和8位的苯甲酸酯是旋转位置,而在NU-901中,这些苯甲酸酯是旋转位置。用相对于the平面分布不均的基团取代pyr的碳2和碳7可以稳定可以形成NU-1000的旋转异构体,因此可以稳定NU-1000本身。我们通过在周期性电子结构计算中应用PBE-D2和M06-L交换相关函数来比较连接体上不同取代基的NU-1000和NU-901的能量,从而证实了这一点。在NU-1000中,与Zr 6配位的苯甲酸酯的苯环节点形成袋状结构。在未取代的NU-1000中,小孔中的口袋较小,并且由于较短的苯-前体距离和更有利的类似分散的相互作用,因此更强地吸引了原子层沉积(ALD)步骤的前驱物,从而导致了小金属中的选择性金属沉积。毛孔。我们研究了取代的NU-1000大孔和小孔中Zr 6节点上Zn(Et)2(ALD前体)的袋大小和吸附能,发现更大的取代基不仅压缩了大孔中的袋,但在大孔中距ALD前体的距离也较短。这两个因素有助于更有利的分散相互作用,并导致较小的金属沉积在小孔中的趋势。
















































 京公网安备 11010802027423号
京公网安备 11010802027423号