当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanism and Origins of Ligand-Controlled Stereoselectivity of Ni-Catalyzed Suzuki–Miyaura Coupling with Benzylic Esters: A Computational Study
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2017-09-07 00:00:00 , DOI: 10.1021/jacs.7b04973 Shuo-Qing Zhang 1 , Buck L. H. Taylor 2, 3 , Chong-Lei Ji 1 , Yuan Gao 1 , Michael R. Harris 4 , Luke E. Hanna 4 , Elizabeth R. Jarvo 4 , K. N. Houk 2 , Xin Hong 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2017-09-07 00:00:00 , DOI: 10.1021/jacs.7b04973 Shuo-Qing Zhang 1 , Buck L. H. Taylor 2, 3 , Chong-Lei Ji 1 , Yuan Gao 1 , Michael R. Harris 4 , Luke E. Hanna 4 , Elizabeth R. Jarvo 4 , K. N. Houk 2 , Xin Hong 1
Affiliation
|
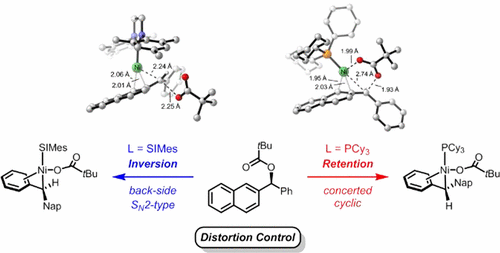
Nickel catalysts have shown unique ligand control of stereoselectivity in the Suzuki–Miyaura cross-coupling of boronates with benzylic pivalates and derivatives involving C(sp3)–O cleavage. The SIMes ligand (1,3-dimesityl-4,5-dihydroimidazol-2-ylidene) produces the stereochemically inverted C–C coupling product, while the tricyclohexylphosphine (PCy3) ligand delivers the retained stereochemistry. We have explored the mechanism and origins of the ligand-controlled stereoselectivity with density functional theory (DFT) calculations. The oxidative addition determines the stereoselectivity with two competing transition states, an SN2 back-side attack type transition state that inverts the benzylic stereogenic center and a concerted oxidative addition through a cyclic transition state, which provides stereoretention. The key difference between the two transition states is the substrate–nickel–ligand angle distortion; the ligand controls the selectivity by differentiating the ease of this angle distortion. For the PCy3 ligand, the nickel–ligand interaction involves mainly σ-donation, which does not require a significant energy penalty for the angle distortion. The facile angle distortion with PCy3 ligand allows the favorable cyclic oxidative addition transition state, leading to the stereoretention. For the SIMes ligand, the extra d–p back-donation from nickel to the coordinating carbene increases the rigidity of the nickel–ligand bond, and the corresponding angle distortion is more difficult. This makes the concerted cyclic oxidative addition unfavorable with SIMes ligand, and the back-side SN2-type oxidative addition delivers the stereoinversion.
中文翻译:

镍催化的铃木-宫浦偶联剂与苯甲酸酯的配体控制的立体选择性机理及成因:计算研究
镍催化剂在硼酸酯与新戊酸苄酯和涉及C(sp 3)-O裂解的衍生物的Suzuki-Miyaura交叉偶联中显示出独特的配体控制立体选择性。SIMes配体(1,3-dimesityl-4,5-dihydroimidazol-2-yylne)产生立体化学反转的C-C偶联产物,而三环己基膦(PCy 3)配体提供保留的立体化学。我们已经通过密度泛函理论(DFT)计算探索了配体控制的立体选择性的机理和起源。氧化加成决定了两个竞争性过渡态S N的立体选择性2个背面攻击型过渡态,其通过循环过渡态使苄基立体发生中心和协同的氧化加成反转,从而提供立体保留。两种过渡态之间的主要区别在于基材-镍-配体角度畸变。配体通过区分该角度畸变的难易程度来控制选择性。对于PCy 3配体,镍-配体相互作用主要涉及σ-给体,它不需要为角畸变带来明显的能量损失。PCy 3的轻松角度失真配体允许有利的环状氧化加成过渡态,导致立体保留。对于SIMes配体,从镍到配位卡宾的额外d-p返配增加了镍-配体键的刚度,并且相应的角度变形也更加困难。这使得协同环状氧化加成不宜用SIMES配体,和背面侧边S Ñ 2型氧化加成递送stereoinversion。
更新日期:2017-09-07
中文翻译:
镍催化的铃木-宫浦偶联剂与苯甲酸酯的配体控制的立体选择性机理及成因:计算研究
镍催化剂在硼酸酯与新戊酸苄酯和涉及C(sp 3)-O裂解的衍生物的Suzuki-Miyaura交叉偶联中显示出独特的配体控制立体选择性。SIMes配体(1,3-dimesityl-4,5-dihydroimidazol-2-yylne)产生立体化学反转的C-C偶联产物,而三环己基膦(PCy 3)配体提供保留的立体化学。我们已经通过密度泛函理论(DFT)计算探索了配体控制的立体选择性的机理和起源。氧化加成决定了两个竞争性过渡态S N的立体选择性2个背面攻击型过渡态,其通过循环过渡态使苄基立体发生中心和协同的氧化加成反转,从而提供立体保留。两种过渡态之间的主要区别在于基材-镍-配体角度畸变。配体通过区分该角度畸变的难易程度来控制选择性。对于PCy 3配体,镍-配体相互作用主要涉及σ-给体,它不需要为角畸变带来明显的能量损失。PCy 3的轻松角度失真配体允许有利的环状氧化加成过渡态,导致立体保留。对于SIMes配体,从镍到配位卡宾的额外d-p返配增加了镍-配体键的刚度,并且相应的角度变形也更加困难。这使得协同环状氧化加成不宜用SIMES配体,和背面侧边S Ñ 2型氧化加成递送stereoinversion。














































 京公网安备 11010802027423号
京公网安备 11010802027423号