当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning Force Field Parameters from Ab Initio Data
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-09-01 00:00:00 , DOI: 10.1021/acs.jctc.7b00521 Ying Li 1 , Hui Li 2 , Frank C. Pickard 3 , Badri Narayanan 4 , Fatih G. Sen 4 , Maria K. Y. Chan 4, 5 , Subramanian K. R. S. Sankaranarayanan 4, 5 , Bernard R. Brooks 3 , Benoît Roux 2, 4, 5
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-09-01 00:00:00 , DOI: 10.1021/acs.jctc.7b00521 Ying Li 1 , Hui Li 2 , Frank C. Pickard 3 , Badri Narayanan 4 , Fatih G. Sen 4 , Maria K. Y. Chan 4, 5 , Subramanian K. R. S. Sankaranarayanan 4, 5 , Bernard R. Brooks 3 , Benoît Roux 2, 4, 5
Affiliation
|
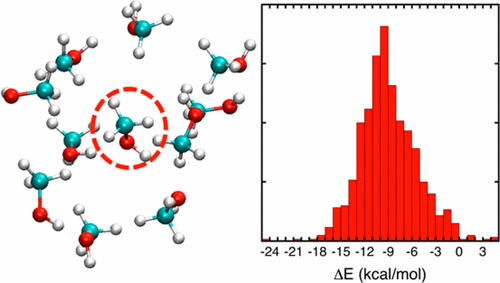
Machine learning (ML) techniques with the genetic algorithm (GA) have been applied to determine a polarizable force field parameters using only ab initio data from quantum mechanics (QM) calculations of molecular clusters at the MP2/6-31G(d,p), DFMP2(fc)/jul-cc-pVDZ, and DFMP2(fc)/jul-cc-pVTZ levels to predict experimental condensed phase properties (i.e., density and heat of vaporization). The performance of this ML/GA approach is demonstrated on 4943 dimer electrostatic potentials and 1250 cluster interaction energies for methanol. Excellent agreement between the training data set from QM calculations and the optimized force field model was achieved. The results were further improved by introducing an offset factor during the machine learning process to compensate for the discrepancy between the QM calculated energy and the energy reproduced by optimized force field, while maintaining the local “shape” of the QM energy surface. Throughout the machine learning process, experimental observables were not involved in the objective function, but were only used for model validation. The best model, optimized from the QM data at the DFMP2(fc)/jul-cc-pVTZ level, appears to perform even better than the original AMOEBA force field (amoeba09.prm), which was optimized empirically to match liquid properties. The present effort shows the possibility of using machine learning techniques to develop descriptive polarizable force field using only QM data. The ML/GA strategy to optimize force fields parameters described here could easily be extended to other molecular systems.
中文翻译:

从头算数据中的机器学习力场参数
已应用具有遗传算法(GA)的机器学习(ML)技术来确定极化力场参数,仅使用从MP2 / 6-31G(d,p)分子簇的量子力学(QM)计算得出的从头算数据,DFMP2(fc)/ jul-cc-pVDZ和DFMP2(fc)/ jul-cc-pVTZ的水平来预测实验的冷凝相性质(即密度和汽化热)。ML / GA方法的性能在甲醇的4943二聚体静电势和1250簇相互作用能上得到了证明。通过QM计算获得的训练数据集与优化的力场模型之间实现了极好的一致性。通过在机器学习过程中引入补偿因子来补偿QM计算出的能量与优化力场所产生的能量之间的差异,同时保持QM能量表面的局部“形状”,从而进一步改善了结果。在整个机器学习过程中,实验可观察值不涉及目标函数,而仅用于模型验证。从DFMP2(fc)/ jul-cc-pVTZ级别的QM数据优化的最佳模型似乎比原始AMOEBA力场(amoeba09.prm)表现更好,后者根据经验进行了优化以匹配液体特性。当前的工作表明了使用机器学习技术仅使用QM数据来开发描述性可极化力场的可能性。
更新日期:2017-09-04
中文翻译:
从头算数据中的机器学习力场参数
已应用具有遗传算法(GA)的机器学习(ML)技术来确定极化力场参数,仅使用从MP2 / 6-31G(d,p)分子簇的量子力学(QM)计算得出的从头算数据,DFMP2(fc)/ jul-cc-pVDZ和DFMP2(fc)/ jul-cc-pVTZ的水平来预测实验的冷凝相性质(即密度和汽化热)。ML / GA方法的性能在甲醇的4943二聚体静电势和1250簇相互作用能上得到了证明。通过QM计算获得的训练数据集与优化的力场模型之间实现了极好的一致性。通过在机器学习过程中引入补偿因子来补偿QM计算出的能量与优化力场所产生的能量之间的差异,同时保持QM能量表面的局部“形状”,从而进一步改善了结果。在整个机器学习过程中,实验可观察值不涉及目标函数,而仅用于模型验证。从DFMP2(fc)/ jul-cc-pVTZ级别的QM数据优化的最佳模型似乎比原始AMOEBA力场(amoeba09.prm)表现更好,后者根据经验进行了优化以匹配液体特性。当前的工作表明了使用机器学习技术仅使用QM数据来开发描述性可极化力场的可能性。






























 京公网安备 11010802027423号
京公网安备 11010802027423号