当前位置:
X-MOL 学术
›
J. Chem. Eng. Data
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics Simulation for the Calibration of the OPLS Force Field Using DFT Derived Partial Charges for the Screening of Alkyl Phosphate Ligands by Studying Structure, Dynamics, and Thermodynamics
Journal of Chemical & Engineering Data ( IF 2.0 ) Pub Date : 2017-07-24 00:00:00 , DOI: 10.1021/acs.jced.7b00096 Arya Das 1, 2 , Pooja Sahu 2, 3 , Sk. Musharaf Ali 2, 3
Journal of Chemical & Engineering Data ( IF 2.0 ) Pub Date : 2017-07-24 00:00:00 , DOI: 10.1021/acs.jced.7b00096 Arya Das 1, 2 , Pooja Sahu 2, 3 , Sk. Musharaf Ali 2, 3
Affiliation
|
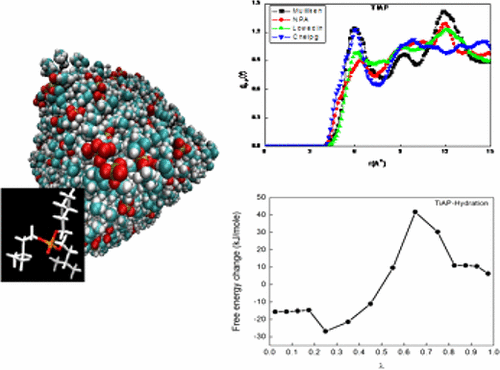
Molecular dynamics (MD) simulations were performed to calibrate the all-atom optimized potential for liquid simulations (OPLS-AA) force field using partial quantum charges calculated from four different population analysis methods: Mulliken, Löwdin, NPA, and ChelpG for predicting the thermophysical properties of pure liquids like tri-n-butylphosphate (TBP), tri-isoamylphosphate (TiAP), triethylphosphate (TEP), and dodecane to determine a potential solvent for the nuclear fuel cycle. The structural, dynamic, and thermodynamic properties were calculated in NVT ensembles by introducing the partial charges on each atom calculated from density functional theory (DFT). The calculated structural and dynamic properties were affected by the different partial charges on TBP, TiAP, and TEP. The estimated liquid density employing partial charges obtained from Mulliken population analysis with OPLS force field leads to an excellent agreement with the experimental data (within 0.36–1.41%). The diffusivity and the pair correlation function (PCF) for all of the ligands have been calculated and validated wherever literature data is available. The free energies of hydration and solvation for all of the ligands were evaluated using thermodynamic integration technique and the hydration free energy for TEP is within 8.3% of the experimental value, and for other properties they are not available in the literature for comparison. Furthermore, the partition coefficient of the ligands calculated using MD derived free energy difference between the water–dodecane system resembles the trend predicted by DFT/COSMO-RS calculations which is in qualitative agreement with the experimental results. Among the four-charge model, the computed dipole moment of TBP and TEP using the Mulliken charge is found to be in good agreement with the experimental results. Finally, the superiority of TiAP over TBP as an extracting agent for the UO22+ ion has been demonstrated by a higher calculated free energy of extraction, ΔGext, over TBP using DFT. Overall the Mulliken charge embedded calibrated OPLS-AA force field is perhaps the most reliable one as it does not incorporate any arbitrary scaling in the force field or Lennard–Jones parameters and thus can be used indubitably to evaluate the liquid state properties of alkyl phosphates and n-alkanes which eventually assist in the invent of future generation extractants.
中文翻译:

通过研究结构,动力学和热力学,使用DFT派生的部分电荷对OPLS力场进行校准的分子动力学模拟,用于筛选烷基磷酸酯配体
使用分子动力学(MD)模拟来校准全原子最优化的液体模拟(OPLS-AA)力场的势能,该分子使用从四种不同的种群分析方法(Mulliken,Löwdin,NPA和ChelpG)计算出的部分量子电荷来预测热物理像三纯液体性质ñ磷酸正丁酯(TBP),磷酸三异戊酯(TiAP),磷酸三乙酯(TEP)和十二烷,以确定核燃料循环的潜在溶剂。通过在每个原子上引入根据密度泛函理论(DFT)计算的部分电荷,可以在NVT集成中计算结构,动力学和热力学性质。TBP,TiAP和TEP上不同的部分电荷会影响计算得到的结构和动力学特性。使用从带有OPLS力场的Mulliken人口分析获得的部分电荷估算的液体密度,导致与实验数据(在0.36-1.41%之内)具有极好的一致性。只要有文献数据,就可以计算和验证所有配体的扩散率和对相关函数(PCF)。使用热力学积分技术评估所有配体的水合和溶剂化自由能,TEP的水合自由能在实验值的8.3%以内,而其他性能在文献中没有可比较的。此外,使用MD得出的水-十二烷体系之间的自由能差计算出的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上吻合。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 实验值的3%,对于其他特性,文献中没有提供它们用于比较。此外,使用MD得出的水-十二烷体系之间的自由能差计算出的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上吻合。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 实验值的3%,对于其他特性,文献中没有提供它们用于比较。此外,使用MD得出的水-十二烷体系之间的自由能差计算出的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上吻合。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 用MD推导的水-十二烷体系之间的自由能差计算的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上是一致的。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 用MD推导的水-十二烷体系之间的自由能差计算的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上是一致的。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂通过使用DFT比TBP提取的更高的计算自由能ΔG ext证明了2 2+离子。总体而言,Mulliken电荷嵌入的经过校准的OPLS-AA力场可能是最可靠的,因为它没有在力场或Lennard-Jones参数中包含任何任意缩放,因此可以毫无疑问地用于评估烷基磷酸酯和最终有助于发明下一代萃取剂的正构烷烃。
更新日期:2017-07-25
中文翻译:
通过研究结构,动力学和热力学,使用DFT派生的部分电荷对OPLS力场进行校准的分子动力学模拟,用于筛选烷基磷酸酯配体
使用分子动力学(MD)模拟来校准全原子最优化的液体模拟(OPLS-AA)力场的势能,该分子使用从四种不同的种群分析方法(Mulliken,Löwdin,NPA和ChelpG)计算出的部分量子电荷来预测热物理像三纯液体性质ñ磷酸正丁酯(TBP),磷酸三异戊酯(TiAP),磷酸三乙酯(TEP)和十二烷,以确定核燃料循环的潜在溶剂。通过在每个原子上引入根据密度泛函理论(DFT)计算的部分电荷,可以在NVT集成中计算结构,动力学和热力学性质。TBP,TiAP和TEP上不同的部分电荷会影响计算得到的结构和动力学特性。使用从带有OPLS力场的Mulliken人口分析获得的部分电荷估算的液体密度,导致与实验数据(在0.36-1.41%之内)具有极好的一致性。只要有文献数据,就可以计算和验证所有配体的扩散率和对相关函数(PCF)。使用热力学积分技术评估所有配体的水合和溶剂化自由能,TEP的水合自由能在实验值的8.3%以内,而其他性能在文献中没有可比较的。此外,使用MD得出的水-十二烷体系之间的自由能差计算出的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上吻合。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 实验值的3%,对于其他特性,文献中没有提供它们用于比较。此外,使用MD得出的水-十二烷体系之间的自由能差计算出的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上吻合。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 实验值的3%,对于其他特性,文献中没有提供它们用于比较。此外,使用MD得出的水-十二烷体系之间的自由能差计算出的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上吻合。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 用MD推导的水-十二烷体系之间的自由能差计算的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上是一致的。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂 用MD推导的水-十二烷体系之间的自由能差计算的配体分配系数类似于DFT / COSMO-RS计算所预测的趋势,与实验结果在质量上是一致的。在四电荷模型中,发现使用Mulliken电荷计算出的TBP和TEP偶极矩与实验结果非常吻合。最后,TiAP优于TBP作为UO的萃取剂通过使用DFT比TBP提取的更高的计算自由能ΔG ext证明了2 2+离子。总体而言,Mulliken电荷嵌入的经过校准的OPLS-AA力场可能是最可靠的,因为它没有在力场或Lennard-Jones参数中包含任何任意缩放,因此可以毫无疑问地用于评估烷基磷酸酯和最终有助于发明下一代萃取剂的正构烷烃。






























 京公网安备 11010802027423号
京公网安备 11010802027423号