当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
迈向基于第一原理的高精度电化学Pourbaix图预测
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2015-07-30 00:00:00 , DOI: 10.1021/acs.jpcc.5b03169
Zhenhua Zeng 1 , Maria K. Y. Chan 2 , Zhi-Jian Zhao 1 , Joseph Kubal 1 , Dingxin Fan 1 , Jeffrey Greeley 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2015-07-30 00:00:00 , DOI: 10.1021/acs.jpcc.5b03169
Zhenhua Zeng 1 , Maria K. Y. Chan 2 , Zhi-Jian Zhao 1 , Joseph Kubal 1 , Dingxin Fan 1 , Jeffrey Greeley 1
Affiliation
|
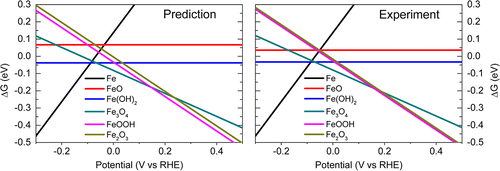
电化学势/ pH(Pourbaix)图是许多水性电化学过程的基础,对于识别从电催化到腐蚀过程的金属稳定相至关重要。尽管标准DFT计算是预测此类图的潜在强大工具,但过渡金属(羟基)氧化物描述中的固有错误以及对范德华相互作用的忽略,即使是最简单的纯正,也限制了此类预测的可靠性。金属块状化合物,以及对更复杂的合金或表面结构的相应预测,甚至更具挑战性。在当前工作中,通过协同使用Hubbard U校正,最新的色散校正以及用于计算的水基体积参考状态,这些误差都会得到系统地校正。该方法在Pourbaix图中描述了某些化合物中含羟基的官能团之间发生的弱结合,纠正了过渡金属化合物中的自相互作用错误,并通过保持参考状态之间一致的氧化态来减少了氧原子上的残留错误,水和相关的本体相。在一系列的块状过渡金属(Mn,Fe,Co和Ni)氢氧化物,羟基氧化物,二元和三元氧化物上显示出强大的性能,其中氧化还原和(脱水)脱水的相应热力学描述为标准误为0.04。每个反应式单位的eV。该方法进一步保留了与电化学有关的本体反应(例如水的形成)的整体热力学的准确描述,这是促进对表面上电化学过程的反应能进行准确分析的必要条件。该方案的总体通用性和可转移性表明,它可在构建广泛的电化学相图(包括用于腐蚀和电催化的大量Pourbaix图和感兴趣的表面相图)的构建中找到有用的应用程序。

"点击查看英文标题和摘要"
更新日期:2015-07-30
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号