当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Thermodynamic Simulation of the RDX–Aluminum Interface Using ReaxFF Molecular Dynamics
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2017-06-28 00:00:00 , DOI: 10.1021/acs.jpcc.7b03108
Ning Wang 1 , Jinhua Peng 1 , Aimin Pang 2 , Tieshan He 2 , Fang Du 2 , Andres Jaramillo-Botero 3
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2017-06-28 00:00:00 , DOI: 10.1021/acs.jpcc.7b03108
Ning Wang 1 , Jinhua Peng 1 , Aimin Pang 2 , Tieshan He 2 , Fang Du 2 , Andres Jaramillo-Botero 3
Affiliation
|
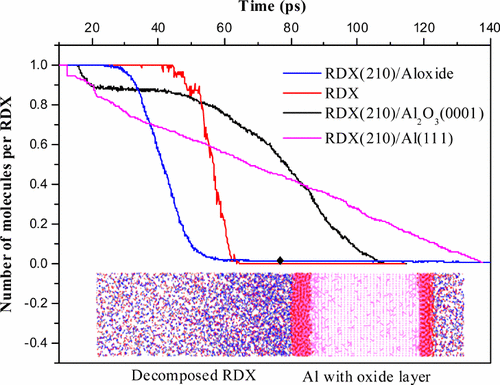
We use reactive molecular dynamics (RMD) simulations to study the interface between cyclotrimethylene trinitramine (RDX) and aluminum (Al) with different oxide layers to elucidate the effect of nanosized Al on thermal decomposition of RDX. A published ReaxFF force field for C/H/N/O elements was retrained to incorporate Al interactions and then used in RMD simulations to characterize compound energetic materials. We find that the predicted adsorption energies for RDX on the Al(111) surface and the apparent activation energies of RDX and RDX/Al are in agreement with ab initio calculations. The Al(111) surface-assisted decomposition of RDX occurs spontaneously without potential barriers, but the decomposition rate becomes slow when compared with that for RDX powder. We also find that the Al(111) surface with an oxide layer (Al oxide) slightly increases the potential barriers for decomposition of RDX molecules, while α-Al2O3(0001) retards thermal decomposition of RDX, due to the changes in thermal decomposition kinetics. The most likely mechanism for the thermal decomposition of RDX powder is described by the Avrami–Erofeev equation, with n = 3/4, as random nucleation and subsequent growth model. Although the decomposition mechanism of RDX molecules in the RDX/Al matrix complies with three-dimensional diffusion, Jander’s equation for RDX(210)/Al oxide and the Zhuralev–Lesokin–Tempelman (Z-L-T) equation for RDX(210)/Al2O3(0001) provide a more accurate description. We conclude that the origin of these differences in dynamic behavior is due to the variations in the oxide layer morphologies.
中文翻译:

使用ReaxFF分子动力学的RDX-铝界面的热力学模拟
我们使用反应分子动力学(RMD)模拟来研究具有不同氧化物层的环三亚甲基三硝胺(RDX)和铝(Al)之间的界面,以阐明纳米尺寸Al对RDX热分解的影响。对已发布的C / H / N / O元素的ReaxFF力场进行了重新训练,以纳入Al相互作用,然后在RMD模拟中用于表征复合含能材料。我们发现,RDX在Al(111)表面上的预测吸附能与RDX和RDX / Al的表观活化能与从头算相符。RDX的Al(111)表面辅助分解是自发发生的,没有势垒,但与RDX粉末相比,分解速率变慢。由于热分解动力学的变化,2 O 3(0001)阻碍了RDX的热分解。为RDX粉末的热分解的最可能的机制是由阿夫拉米-Erofeev方程说明的那样,Ñ = 3 / 4,作为随机的成核和随后的生长模式。尽管RDX / Al矩阵中RDX分子的分解机理符合三维扩散,但RDX(210)/ Al氧化物的Jander方程和RDX(210)/ Al 2 O的Zhuralev-Lesokin-Tempelman(ZLT)方程3(0001)提供更准确的描述。我们得出结论,这些动态行为差异的根源是由于氧化物层形态的变化。
更新日期:2017-06-29
中文翻译:
使用ReaxFF分子动力学的RDX-铝界面的热力学模拟
我们使用反应分子动力学(RMD)模拟来研究具有不同氧化物层的环三亚甲基三硝胺(RDX)和铝(Al)之间的界面,以阐明纳米尺寸Al对RDX热分解的影响。对已发布的C / H / N / O元素的ReaxFF力场进行了重新训练,以纳入Al相互作用,然后在RMD模拟中用于表征复合含能材料。我们发现,RDX在Al(111)表面上的预测吸附能与RDX和RDX / Al的表观活化能与从头算相符。RDX的Al(111)表面辅助分解是自发发生的,没有势垒,但与RDX粉末相比,分解速率变慢。由于热分解动力学的变化,2 O 3(0001)阻碍了RDX的热分解。为RDX粉末的热分解的最可能的机制是由阿夫拉米-Erofeev方程说明的那样,Ñ = 3 / 4,作为随机的成核和随后的生长模式。尽管RDX / Al矩阵中RDX分子的分解机理符合三维扩散,但RDX(210)/ Al氧化物的Jander方程和RDX(210)/ Al 2 O的Zhuralev-Lesokin-Tempelman(ZLT)方程3(0001)提供更准确的描述。我们得出结论,这些动态行为差异的根源是由于氧化物层形态的变化。
































 京公网安备 11010802027423号
京公网安备 11010802027423号