当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab Initio Adsorption Isotherms for Molecules with Lateral Interactions: CO2 in Metal–Organic Frameworks
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2017-06-06 00:00:00 , DOI: 10.1021/acs.jpcc.7b02806 Kaido Sillar 1, 2 , Arpan Kundu 1 , Joachim Sauer 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2017-06-06 00:00:00 , DOI: 10.1021/acs.jpcc.7b02806 Kaido Sillar 1, 2 , Arpan Kundu 1 , Joachim Sauer 1
Affiliation
|
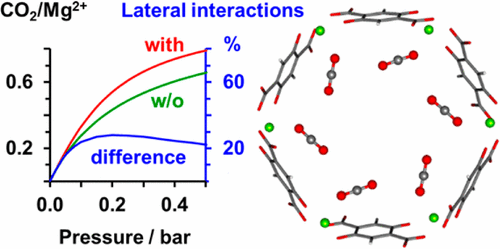
Adsorption of carbon dioxide in the metal–organic framework CPO-27-Mg (Mg-MOF-74) is examined. We use accurate quantum chemical ab initio methods (wave function-type electron correlation methods for cluster models combined with density functional theory for periodic systems) to calculate gas–surface site and gas–gas interactions. At 298 K, the “zero-coverage” enthalpy and Gibbs free energy of CO2 adsorption on Mg2+ sites are −46 and −9 kJ/mol, respectively; for linker sites these values are −30 and +5 kJ/mol, respectively. For full monolayer coverage lateral interactions from nearby molecules contribute −6 and −5 kJ/mol to the adsorption enthalpy for CO2 at Mg2+ and linker sites, respectively. The predicted heats of adsorption and free energies of adsorption agree within 2.6 and 0.8 kJ/mol, respectively, with experimental values well within chemical accuracy limits (4.2 kJ/mol). We use two different ways of calculating isotherms from equilibrium constants for individual sites and interaction energies: (i) a Langmuir model, augmented with the mean-field (MF) approximation for lateral interactions, and (ii) grand canonical Monte Carlo (GCMC) simulations on a lattice of sites, which agree very well with each other. We use GCMC data to examine how different isotherm models (Langmuir, dual-site Langmuir, Sips, Toth, and mean-field) fit them. We conclude that the MF model yields the best fit over a wide pressure range with physically meaningful parameters, i.e., adsorption constants for individual sites and lateral interaction energies.
中文翻译:

具有横向相互作用的分子的从头算起吸附等温线:金属-有机骨架中的CO 2
研究了金属-有机骨架CPO-27-Mg(Mg-MOF-74)中二氧化碳的吸附。我们使用精确的量子化学从头算方法(簇模型的波函数型电子相关方法,与周期性系统的密度泛函理论相结合)来计算气-液面和气-气相互作用。在298 K时,Mg 2+位置上的CO 2的“零覆盖”焓和吉布斯自由能分别为-46和-9 kJ / mol。对于接头位点,这些值分别为-30和+5 kJ / mol。对于完全的单层覆盖,附近分子的横向相互作用对Mg 2+处的CO 2吸附焓贡献了-6和-5 kJ / mol。和链接器站点。预测的吸附热和吸附自由能分别在2.6 kJ / mol和0.8 kJ / mol之间,实验值完全在化学精度极限(4.2 kJ / mol)之内。我们使用两种不同的方法根据各个位点的平衡常数和相互作用能来计算等温线:(i)Langmuir模型,通过横向相互作用的均值场(MF)近似进行增强,以及(ii)大规范蒙特卡洛(GCMC)站点格上的模拟,它们彼此非常吻合。我们使用GCMC数据来检查不同的等温线模型(Langmuir,双站点Langmuir,Sips,Toth和均值场)如何拟合它们。我们得出的结论是,MF模型在具有物理意义的参数(即,
更新日期:2017-06-06
中文翻译:
具有横向相互作用的分子的从头算起吸附等温线:金属-有机骨架中的CO 2
研究了金属-有机骨架CPO-27-Mg(Mg-MOF-74)中二氧化碳的吸附。我们使用精确的量子化学从头算方法(簇模型的波函数型电子相关方法,与周期性系统的密度泛函理论相结合)来计算气-液面和气-气相互作用。在298 K时,Mg 2+位置上的CO 2的“零覆盖”焓和吉布斯自由能分别为-46和-9 kJ / mol。对于接头位点,这些值分别为-30和+5 kJ / mol。对于完全的单层覆盖,附近分子的横向相互作用对Mg 2+处的CO 2吸附焓贡献了-6和-5 kJ / mol。和链接器站点。预测的吸附热和吸附自由能分别在2.6 kJ / mol和0.8 kJ / mol之间,实验值完全在化学精度极限(4.2 kJ / mol)之内。我们使用两种不同的方法根据各个位点的平衡常数和相互作用能来计算等温线:(i)Langmuir模型,通过横向相互作用的均值场(MF)近似进行增强,以及(ii)大规范蒙特卡洛(GCMC)站点格上的模拟,它们彼此非常吻合。我们使用GCMC数据来检查不同的等温线模型(Langmuir,双站点Langmuir,Sips,Toth和均值场)如何拟合它们。我们得出的结论是,MF模型在具有物理意义的参数(即,






























 京公网安备 11010802027423号
京公网安备 11010802027423号