当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Improved Polarizable Dipole–Dipole Interaction Model for Hydrogen Bonding, Stacking, T-Shaped, and X–H···π Interactions
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-05-10 00:00:00 , DOI: 10.1021/acs.jctc.6b00936 Xi-Chan Gao 1 , Qiang Hao 1 , Chang-Sheng Wang 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-05-10 00:00:00 , DOI: 10.1021/acs.jctc.6b00936 Xi-Chan Gao 1 , Qiang Hao 1 , Chang-Sheng Wang 1
Affiliation
|
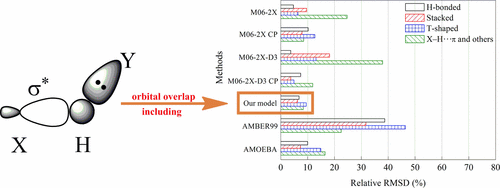
The polarizable dipole–dipole interaction model was formulated in our laboratory to rapidly simulate hydrogen bonding in biosystems. In this paper, this model is improved and further parametrized for stacking, T-shaped, and X–H···π interactions by adding the orbital overlap term and fitting to 19 CCSD(T)/CBS interaction energy curves of training dimers. The performance of our model is assessed through its application to more than 100 complexes, including hydrogen-bonded, stacked, T-shaped, and X–H···π complexes. For 124 relatively small testing complexes, our model reproduces benchmark equilibrium intermolecular distances with a root-mean-square deviation (RMSD) of 0.08 Å, and it reproduces benchmark interaction energies with a 0.64 kcal/mol RMSD. For 14 large noncovalent complexes, our model reproduces benchmark equilibrium intermolecular distances with a RMSD of 0.05 Å, and it reproduces benchmark interaction energies with a 0.80 kcal/mol RMSD. Extensive comparisons are made to interaction energies calculated via the M06-2X and M06-2X-D3 methods, via the well-known nonpolarizable AMBER99 force field method, via the popular polarizable AMOEBA force field method, and via semiempirical quantum mechanical (SQM) methods. Our statistical evaluations show that our model outperforms the AMBER99, AMOEBA, and SQM methods and is as accurate as the M06-2X and M06-2X-D3 methods. In summary, the model developed in this work is reasonable, and the newly introduced orbital overlap term is effective in the accurate modeling of the noncovalent interactions. Our testing results also indicate that the polarization interaction term is important in the evaluation of hydrogen bonding, whereas the orbital overlap is important in examining short hydrogen bonding, T-shaped, and X–H···π interactions. Our model may serve as a new tool for modeling biological systems where hydrogen bonding, stacking, T-shaped, and X–H···π interactions are of general importance.
中文翻译:

氢键键合,堆积,T形和X–H···π相互作用的改进的极化极化偶极-偶极相互作用模型
在我们的实验室中建立了可极化的偶极-偶极相互作用模型,以快速模拟生物系统中的氢键。在本文中,通过添加轨道重叠项并将其拟合到训练二聚体的19 CCSD(T)/ CBS相互作用能曲线,对该模型进行了改进和参数化,以用于堆叠,T形和X–H··π相互作用。通过将其应用于100多个配合物,包括氢键,堆积,T形和X–H··π配合物,可以评估我们模型的性能。对于124个相对较小的测试复合物,我们的模型再现了均方根偏差(RMSD)为0.08Å的基准平衡分子间距离,并再现了0.64 kcal / mol RMSD的基准相互作用能。对于14种大型非共价复合物,我们的模型以0.05Å的RMSD再现了基准平衡分子间距离,并且以0.80 kcal / mol的RMSD再现了基准相互作用能。广泛比较了通过M06-2X和M06-2X-D3方法,通过众所周知的非极化AMBER99力场方法,通过流行的极化AMOEBA力场方法以及通过半经验量子力学(SQM)方法计算出的相互作用能。我们的统计评估表明,我们的模型优于AMBER99,AMOEBA和SQM方法,并且与M06-2X和M06-2X-D3方法一样准确。总之,在这项工作中开发的模型是合理的,并且新引入的轨道重叠项对于非共价相互作用的精确建模是有效的。我们的测试结果还表明,极化相互作用项对氢键的评估很重要,而轨道重叠对检查短氢键,T形和X–H··π相互作用很重要。我们的模型可以作为对生物系统建模的新工具,其中氢键,堆积,T形和X–H··π相互作用至关重要。
更新日期:2017-05-20
中文翻译:
氢键键合,堆积,T形和X–H···π相互作用的改进的极化极化偶极-偶极相互作用模型
在我们的实验室中建立了可极化的偶极-偶极相互作用模型,以快速模拟生物系统中的氢键。在本文中,通过添加轨道重叠项并将其拟合到训练二聚体的19 CCSD(T)/ CBS相互作用能曲线,对该模型进行了改进和参数化,以用于堆叠,T形和X–H··π相互作用。通过将其应用于100多个配合物,包括氢键,堆积,T形和X–H··π配合物,可以评估我们模型的性能。对于124个相对较小的测试复合物,我们的模型再现了均方根偏差(RMSD)为0.08Å的基准平衡分子间距离,并再现了0.64 kcal / mol RMSD的基准相互作用能。对于14种大型非共价复合物,我们的模型以0.05Å的RMSD再现了基准平衡分子间距离,并且以0.80 kcal / mol的RMSD再现了基准相互作用能。广泛比较了通过M06-2X和M06-2X-D3方法,通过众所周知的非极化AMBER99力场方法,通过流行的极化AMOEBA力场方法以及通过半经验量子力学(SQM)方法计算出的相互作用能。我们的统计评估表明,我们的模型优于AMBER99,AMOEBA和SQM方法,并且与M06-2X和M06-2X-D3方法一样准确。总之,在这项工作中开发的模型是合理的,并且新引入的轨道重叠项对于非共价相互作用的精确建模是有效的。我们的测试结果还表明,极化相互作用项对氢键的评估很重要,而轨道重叠对检查短氢键,T形和X–H··π相互作用很重要。我们的模型可以作为对生物系统建模的新工具,其中氢键,堆积,T形和X–H··π相互作用至关重要。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号