当前位置:
X-MOL 学术
›
J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Discovery of CREBBP Bromodomain Inhibitors by High-Throughput Docking and Hit Optimization Guided by Molecular Dynamics
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2015-07-15 00:00:00 , DOI: 10.1021/acs.jmedchem.5b00171 Min Xu 1 , Andrea Unzue 2 , Jing Dong 1 , Dimitrios Spiliotopoulos 1 , Cristina Nevado 2 , Amedeo Caflisch 1
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2015-07-15 00:00:00 , DOI: 10.1021/acs.jmedchem.5b00171 Min Xu 1 , Andrea Unzue 2 , Jing Dong 1 , Dimitrios Spiliotopoulos 1 , Cristina Nevado 2 , Amedeo Caflisch 1
Affiliation
|
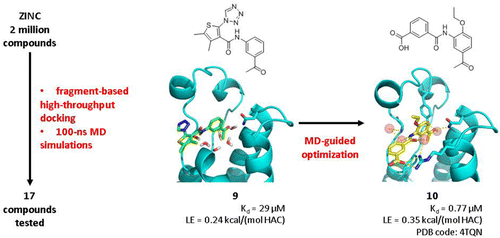
We have identified two chemotypes of CREBBP bromodomain ligands by fragment-based high-throughput docking. Only 17 molecules from the original library of two-million compounds were tested in vitro. Optimization of the two low-micromolar hits, the 4-acylpyrrole 1 and acylbenzene 9, was driven by molecular dynamics results which suggested improvement of the polar interactions with the Arg1173 side chain at the rim of the binding site. The synthesis of only two derivatives of 1 yielded the 4-acylpyrrole 6 which shows a single-digit micromolar affinity for the CREBBP bromodomain and a ligand efficiency of 0.34 kcal/mol per non-hydrogen atom. Optimization of the acylbenzene hit 9 resulted in a series of derivatives with nanomolar potencies, good ligand efficiency and selectivity (see Unzue, A.; Xu, M.; Dong, J.; Wiedmer, L.; Spiliotopoulos, D.; Caflisch, A.; Nevado, C. Fragment-Based Design of Selective Nanomolar Ligands of the CREBBP Bromodomain. J. Med. Chem. 2015, DOI: 10.1021/acs.jmedchem.5b00172). The in silico predicted binding mode of the acylbenzene derivative 10 was validated by solving the structure of the complex with the CREBBP bromodomain.
中文翻译:

高通量对接发现CREBBP溴结构域抑制剂,分子动力学指导命中优化
我们已经通过基于片段的高通量对接确定了两种CREBBP溴结构域配体的化学型。在体外仅对来自200万个化合物的原始库中的17个分子进行了测试。分子动力学结果驱动了两个低微摩尔分子命中率的优化,即4-酰基吡咯1和酰基苯9,这表明结合位点边缘与Arg1173侧链的极性相互作用得到改善。仅两种衍生物1的合成产生了4-酰基吡咯6,其对CREBBP溴结构域表现出一位数的微摩尔亲和力,并且每个非氢原子的配体效率为0.34kcal / mol。酰基苯的优化命中9 产生了一系列具有纳摩尔浓度,良好的配体效率和选择性的衍生物(请参阅恩祖(A. 徐敏 董建 威德默(Wedmer)Spiliotopoulos,D。Caflisch,A .;内华达州C. CREBBP溴结构域的选择性纳摩尔配体的基于片段的设计。J. Med。化学 2015年,DOI:10.1021 / acs.jmedchem.5b00172)。通过解析具有CREBBP溴结构域的复合物的结构来验证酰基苯衍生物10的计算机模拟预测结合模式。
更新日期:2015-07-15
中文翻译:
高通量对接发现CREBBP溴结构域抑制剂,分子动力学指导命中优化
我们已经通过基于片段的高通量对接确定了两种CREBBP溴结构域配体的化学型。在体外仅对来自200万个化合物的原始库中的17个分子进行了测试。分子动力学结果驱动了两个低微摩尔分子命中率的优化,即4-酰基吡咯1和酰基苯9,这表明结合位点边缘与Arg1173侧链的极性相互作用得到改善。仅两种衍生物1的合成产生了4-酰基吡咯6,其对CREBBP溴结构域表现出一位数的微摩尔亲和力,并且每个非氢原子的配体效率为0.34kcal / mol。酰基苯的优化命中9 产生了一系列具有纳摩尔浓度,良好的配体效率和选择性的衍生物(请参阅
































 京公网安备 11010802027423号
京公网安备 11010802027423号