Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Charge-Transfer-Driven Nonplanar Adsorption of F4TCNQ Molecules on Epitaxial Graphene
ACS Nano ( IF 15.8 ) Pub Date : 2017-05-03 00:00:00 , DOI: 10.1021/acsnano.7b01599 Avijit Kumar 1 , Kaustuv Banerjee 1 , Marc Dvorak 1 , Fabian Schulz 1 , Ari Harju 1 , Patrick Rinke 1 , Peter Liljeroth 1
ACS Nano ( IF 15.8 ) Pub Date : 2017-05-03 00:00:00 , DOI: 10.1021/acsnano.7b01599 Avijit Kumar 1 , Kaustuv Banerjee 1 , Marc Dvorak 1 , Fabian Schulz 1 , Ari Harju 1 , Patrick Rinke 1 , Peter Liljeroth 1
Affiliation
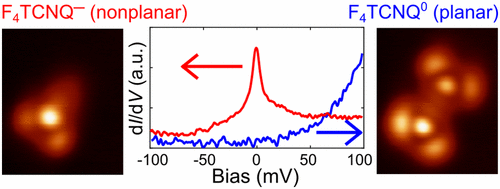
|
π-conjugated organic molecules tend to adsorb in a planar configuration on graphene irrespective of their charge state. In contrast, here we demonstrate charging-induced strong structural relaxation of tetrafluorotetracyanoquinodimethane (F4TCNQ) on epitaxial graphene on Ir(111) (G/Ir(111)). The work function modulation over the graphene moiré unit cell causes site-selective charging of F4TCNQ. Upon charging, the molecule anchors to the face-centered cubic sites of the G/Ir(111) moiré through one or two cyano groups. The reaction is reversible and can be triggered on a single molecule by moving it between different adsorption sites. We introduce a model taking into account the trade-off between tilt-induced charging and reduced van der Waals interactions, which provides a general framework for understanding charging-induced structural relaxation on weakly interacting substrates. In addition, we argue that the partial sp3 rehybridization of the underlying graphene and the possible bonding mechanism between the cyano groups and the graphene substrate are also relevant for the complete understanding of the experiments. These results provide insight into molecular charging on graphene, and they are directly relevant for potential device applications where the use of molecules has been suggested for doping and band structure engineering.
中文翻译:

F 4 TCNQ分子在外延石墨烯上的电荷转移驱动非平面吸附
π共轭有机分子不考虑其电荷状态而倾向于以平面构型吸附在石墨烯上。相比之下,在这里我们展示了在Ir(111)(G / Ir(111))上外延石墨烯上的四氟四氰基喹二甲烷(F 4 TCNQ)的电荷诱导的强结构弛豫。石墨烯莫尔条纹晶胞上的功函数调制导致F 4的位点选择性带电TCNQ。充电后,分子通过一个或两个氰基锚定到G / Ir(111)波纹的面心立方位置。该反应是可逆的,并且可以通过在不同的吸附位点之间移动来触发单个分子。我们介绍了一种模型,该模型考虑了倾斜诱导的电荷与减少的范德华相互作用之间的权衡,该模型为理解弱相互作用的基底上的电荷诱导的结构弛豫提供了一个通用框架。此外,我们认为部分sp 3底层石墨烯的再杂交以及氰基与石墨烯底物之间可能的键合机理也与对实验的全面理解有关。这些结果提供了对石墨烯上分子带电的洞察力,并且与潜在的器件应用直接相关,在这种应用中,已建议使用分子进行掺杂和能带结构工程。
更新日期:2017-05-10
中文翻译:
F 4 TCNQ分子在外延石墨烯上的电荷转移驱动非平面吸附
π共轭有机分子不考虑其电荷状态而倾向于以平面构型吸附在石墨烯上。相比之下,在这里我们展示了在Ir(111)(G / Ir(111))上外延石墨烯上的四氟四氰基喹二甲烷(F 4 TCNQ)的电荷诱导的强结构弛豫。石墨烯莫尔条纹晶胞上的功函数调制导致F 4的位点选择性带电TCNQ。充电后,分子通过一个或两个氰基锚定到G / Ir(111)波纹的面心立方位置。该反应是可逆的,并且可以通过在不同的吸附位点之间移动来触发单个分子。我们介绍了一种模型,该模型考虑了倾斜诱导的电荷与减少的范德华相互作用之间的权衡,该模型为理解弱相互作用的基底上的电荷诱导的结构弛豫提供了一个通用框架。此外,我们认为部分sp 3底层石墨烯的再杂交以及氰基与石墨烯底物之间可能的键合机理也与对实验的全面理解有关。这些结果提供了对石墨烯上分子带电的洞察力,并且与潜在的器件应用直接相关,在这种应用中,已建议使用分子进行掺杂和能带结构工程。
































 京公网安备 11010802027423号
京公网安备 11010802027423号