当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accelerated Design of Dual-Metal-Site Catalysts via Machine-Learning Prediction
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2025-01-31 , DOI: 10.1021/acs.jpclett.5c00126
Yang Wang, Qian Wang, Xijun Wang, Jing Yang, Jun Jiang, Chuanyi Jia
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2025-01-31 , DOI: 10.1021/acs.jpclett.5c00126
Yang Wang, Qian Wang, Xijun Wang, Jing Yang, Jun Jiang, Chuanyi Jia
|
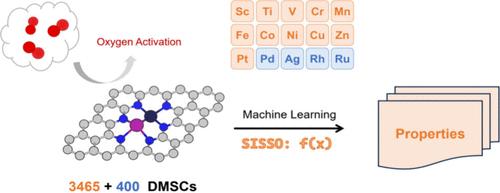
Dual-metal site catalysts (DMSCs) supported on nitrogen-doped graphene have shown great potential in heterogeneous catalysis due to their unique properties and enhanced efficiency. However, the precise control and stabilization of metal dimers, particularly in oxygen activation reactions, present significant challenges in practical applications. In this study, we integrate high-throughput density functional theory calculations with machine learning techniques to predict and optimize the catalytic properties of DMSCs. Transfer learning is employed to enhance the model’s generalization capability, successfully predicting catalytic performance across new metal combinations. Additionally, the application of the SISSO method enables the derivation of interpretable symbolic regression models, revealing critical correlations between electronic structure features and catalytic efficiency. This approach not only advances the understanding of dual-metal site catalysis but also provides a novel framework for the systematic design and optimization of highly efficient catalysts, with broad applicability in catalytic science.
中文翻译:

通过机器学习预测加速双金属位催化剂的设计
由于其独特的性能和更高的效率,负载在氮掺杂石墨烯上的双金属位催化剂 (DMSC) 在多相催化中显示出巨大的潜力。然而,金属二聚体的精确控制和稳定,特别是在氧活化反应中,在实际应用中提出了重大挑战。在这项研究中,我们将高通量密度泛函理论计算与机器学习技术相结合,以预测和优化 DMSC 的催化特性。采用迁移学习来增强模型的泛化能力,成功预测新金属组合的催化性能。此外,SISSO 方法的应用能够推导出可解释的符号回归模型,揭示电子结构特征与催化效率之间的关键相关性。这种方法不仅促进了对双金属位点催化的理解,还为高效催化剂的系统设计和优化提供了新的框架,在催化科学中具有广泛的适用性。
更新日期:2025-01-31
中文翻译:
通过机器学习预测加速双金属位催化剂的设计
由于其独特的性能和更高的效率,负载在氮掺杂石墨烯上的双金属位催化剂 (DMSC) 在多相催化中显示出巨大的潜力。然而,金属二聚体的精确控制和稳定,特别是在氧活化反应中,在实际应用中提出了重大挑战。在这项研究中,我们将高通量密度泛函理论计算与机器学习技术相结合,以预测和优化 DMSC 的催化特性。采用迁移学习来增强模型的泛化能力,成功预测新金属组合的催化性能。此外,SISSO 方法的应用能够推导出可解释的符号回归模型,揭示电子结构特征与催化效率之间的关键相关性。这种方法不仅促进了对双金属位点催化的理解,还为高效催化剂的系统设计和优化提供了新的框架,在催化科学中具有广泛的适用性。
































 京公网安备 11010802027423号
京公网安备 11010802027423号