当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning Prediction of Physicochemical Properties in Lithium‐Ion Battery Electrolytes With Active Learning Applied to Graph Neural Networks
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-12-26 , DOI: 10.1002/jcc.70009 Debojyoti Das, Debdutta Chakraborty
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-12-26 , DOI: 10.1002/jcc.70009 Debojyoti Das, Debdutta Chakraborty
|
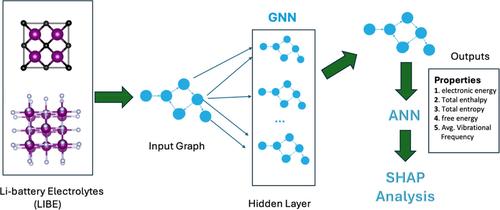
Accurate prediction of physicochemical properties, such as electronic energy, enthalpy, free energy, and average vibrational frequencies, is critical for optimizing lithium‐ion battery (LIB) performance. Traditional methods like density functional theory (DFT) are computationally expensive and inefficient for large‐scale screening. In this study, we apply active learning on top of graph neural networks (GNNs) to efficiently predict these properties. By focusing on uncertain data points, active learning reduces training data size while maintaining high accuracy. Applied to the LIBE and MPcules datasets, the model achieved an R‐squared (R 2 ) values of 0.9977 with a mean absolute error (MAE) of 9.66 Ha for electronic energy and an R 2 values of 0.957 with an MAE of 13.94 cm−1 for average vibrational frequencies. SHapley Additive exPlanations (SHAP) provided insights into key features influencing predictions, such as atomic number and spin multiplicity. This approach enhances both predictive accuracy and model interpretability, offering a scalable solution for LIB electrolyte discovery.
中文翻译:

将主动学习应用于图神经网络的锂离子电池电解质物理化学性质的机器学习预测
准确预测物理化学性质,例如电子能、焓、自由能和平均振动频率,对于优化锂离子电池 (LIB) 性能至关重要。密度泛函理论 (DFT) 等传统方法计算成本高昂且对于大规模筛选效率低下。在这项研究中,我们在图神经网络 (GNN) 之上应用主动学习来有效地预测这些特性。通过关注不确定的数据点,主动学习可以减小训练数据大小,同时保持高精度。应用于 LIBE 和 MPcules 数据集,该模型实现了 0.9977 的 R 平方 (R2) 值,电子能量的平均绝对误差 (MAE) 为 9.66 Ha,R2 值为 0.957,平均振动频率的 MAE 为 13.94 cm-1。SHapley 加法解释 (SHAP) 提供了对影响预测的关键特征的见解,例如原子序数和自旋多重性。这种方法提高了预测准确性和模型可解释性,为 LIB 电解质发现提供了可扩展的解决方案。
更新日期:2024-12-26
中文翻译:
将主动学习应用于图神经网络的锂离子电池电解质物理化学性质的机器学习预测
准确预测物理化学性质,例如电子能、焓、自由能和平均振动频率,对于优化锂离子电池 (LIB) 性能至关重要。密度泛函理论 (DFT) 等传统方法计算成本高昂且对于大规模筛选效率低下。在这项研究中,我们在图神经网络 (GNN) 之上应用主动学习来有效地预测这些特性。通过关注不确定的数据点,主动学习可以减小训练数据大小,同时保持高精度。应用于 LIBE 和 MPcules 数据集,该模型实现了 0.9977 的 R 平方 (R2) 值,电子能量的平均绝对误差 (MAE) 为 9.66 Ha,R2 值为 0.957,平均振动频率的 MAE 为 13.94 cm-1。SHapley 加法解释 (SHAP) 提供了对影响预测的关键特征的见解,例如原子序数和自旋多重性。这种方法提高了预测准确性和模型可解释性,为 LIB 电解质发现提供了可扩展的解决方案。






























 京公网安备 11010802027423号
京公网安备 11010802027423号