npj Computational Materials ( IF 9.4 ) Pub Date : 2024-12-19 , DOI: 10.1038/s41524-024-01493-2 Duo Zhang, Xinzijian Liu, Xiangyu Zhang, Chengqian Zhang, Chun Cai, Hangrui Bi, Yiming Du, Xuejian Qin, Anyang Peng, Jiameng Huang, Bowen Li, Yifan Shan, Jinzhe Zeng, Yuzhi Zhang, Siyuan Liu, Yifan Li, Junhan Chang, Xinyan Wang, Shuo Zhou, Jianchuan Liu, Xiaoshan Luo, Zhenyu Wang, Wanrun Jiang, Jing Wu, Yudi Yang, Jiyuan Yang, Manyi Yang, Fu-Qiang Gong, Linshuang Zhang, Mengchao Shi, Fu-Zhi Dai, Darrin M. York, Shi Liu, Tong Zhu, Zhicheng Zhong, Jian Lv, Jun Cheng, Weile Jia, Mohan Chen, Guolin Ke, Weinan E, Linfeng Zhang, Han Wang
|
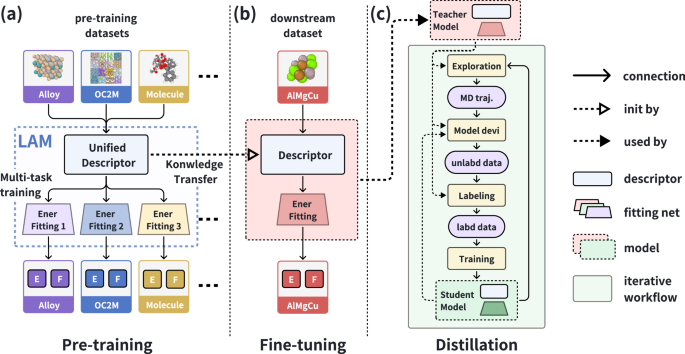
The rapid advancements in artificial intelligence (AI) are catalyzing transformative changes in atomic modeling, simulation, and design. AI-driven potential energy models have demonstrated the capability to conduct large-scale, long-duration simulations with the accuracy of ab initio electronic structure methods. However, the model generation process remains a bottleneck for large-scale applications. We propose a shift towards a model-centric ecosystem, wherein a large atomic model (LAM), pre-trained across multiple disciplines, can be efficiently fine-tuned and distilled for various downstream tasks, thereby establishing a new framework for molecular modeling. In this study, we introduce the DPA-2 architecture as a prototype for LAMs. Pre-trained on a diverse array of chemical and materials systems using a multi-task approach, DPA-2 demonstrates superior generalization capabilities across multiple downstream tasks compared to the traditional single-task pre-training and fine-tuning methodologies. Our approach sets the stage for the development and broad application of LAMs in molecular and materials simulation research.





























 京公网安备 11010802027423号
京公网安备 11010802027423号