当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Vibrational and Structural Properties of PTCDA on Ag(110)
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-12-19 , DOI: 10.1021/acs.jpcc.4c07113 R. Priya, Peter Jakob
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2024-12-19 , DOI: 10.1021/acs.jpcc.4c07113 R. Priya, Peter Jakob
|
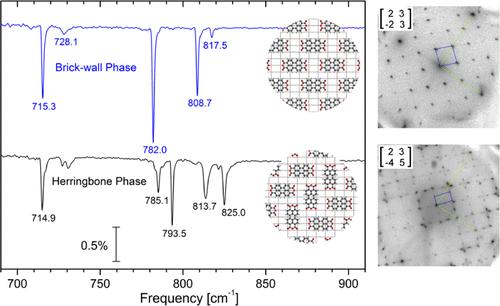
The structure and vibrational properties of 3,4,9,10-perylenetetracarboxylic dianhydride (PTCDA) molecular layers have been investigated on the nonhexagonal Ag(110) substrate. Based on their distinct and unambiguous vibrational signatures, the existing brick wall (BW) and herringbone (HB) phases have been identified and discriminated using infrared absorption spectroscopy. Their particular long-range ordering, e.g., their authenticity, has been verified using spot-profile analysis low energy electron diffraction (SPA-LEED). At variance with earlier studies, we provide conclusive evidence for a commensurate HB structure. Due to rather strong molecule–metal and intermolecular interactions, the transition between the BW and the more dense HB phase proceeds only partially, and we have developed recipes to produce uniform layers comprising either of the two long-range ordered phases. Comparison of PTCDA adsorption on Ag(110), Ag(111), and Au(111) substrates reveals different scenarios regarding interfacial dynamic charge transfer (IDCT) associated with the excitation of vibrational modes. Specifically, the effect of strong, medium strong, and weak interactions with the substrate, in conjunction with chemical bond formation, has been examined regarding the extent of IDCT and the asymmetries of the vibrational line shapes. Our findings are supported by reports in the literature analyzing the location of the former LUMO with respect to the (substrate) Fermi energy as well as the vertical bonding distances and molecular distortions of the PTCDA molecular plane.
中文翻译:

PTCDA 对 Ag 的振动和结构特性 (110)
在非六方 Ag(110) 衬底上研究了 3,4,9,10-苝四羧酸二酐 (PTCDA) 分子层的结构和振动特性。根据它们独特而明确的振动特征,现有的砖墙 (BW) 和人字形 (HB) 相已使用红外吸收光谱进行识别和区分。它们特定的长程排序,例如其真实性,已使用光斑剖面分析低能电子衍射 (SPA-LEED) 进行了验证。与早期研究不同,我们为相应的 HB 结构提供了确凿的证据。由于相当强的分子-金属和分子间相互作用,BW 和更密集的 HB 相之间的转变仅部分进行,并且我们开发了产生由两个长程有序相中的任何一个组成的均匀层的配方。PTCDA 在 Ag(110)、Ag(111) 和 Au(111) 衬底上的吸附比较揭示了与振动模式激发相关的界面动态电荷转移 (IDCT) 的不同情况。具体来说,已经研究了 IDCT 的程度和振动线形状的不对称性,与基材的强、中强和弱相互作用以及化学键形成的影响。我们的发现得到了文献报告的支持,这些文献分析了前 LUMO 相对于(衬底)费米能量的位置以及 PTCDA 分子平面的垂直键合距离和分子畸变。
更新日期:2024-12-19
中文翻译:
PTCDA 对 Ag 的振动和结构特性 (110)
在非六方 Ag(110) 衬底上研究了 3,4,9,10-苝四羧酸二酐 (PTCDA) 分子层的结构和振动特性。根据它们独特而明确的振动特征,现有的砖墙 (BW) 和人字形 (HB) 相已使用红外吸收光谱进行识别和区分。它们特定的长程排序,例如其真实性,已使用光斑剖面分析低能电子衍射 (SPA-LEED) 进行了验证。与早期研究不同,我们为相应的 HB 结构提供了确凿的证据。由于相当强的分子-金属和分子间相互作用,BW 和更密集的 HB 相之间的转变仅部分进行,并且我们开发了产生由两个长程有序相中的任何一个组成的均匀层的配方。PTCDA 在 Ag(110)、Ag(111) 和 Au(111) 衬底上的吸附比较揭示了与振动模式激发相关的界面动态电荷转移 (IDCT) 的不同情况。具体来说,已经研究了 IDCT 的程度和振动线形状的不对称性,与基材的强、中强和弱相互作用以及化学键形成的影响。我们的发现得到了文献报告的支持,这些文献分析了前 LUMO 相对于(衬底)费米能量的位置以及 PTCDA 分子平面的垂直键合距离和分子畸变。





























 京公网安备 11010802027423号
京公网安备 11010802027423号