当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Decoding the Role of Adsorbates Entropy in the Reactivity of Single-Atom Catalysts
ACS Catalysis ( IF 11.3 ) Pub Date : 2024-12-18 , DOI: 10.1021/acscatal.4c04472 Elena Di Simone, Gianvito Vilé, Giovanni Di Liberto, Gianfranco Pacchioni
ACS Catalysis ( IF 11.3 ) Pub Date : 2024-12-18 , DOI: 10.1021/acscatal.4c04472 Elena Di Simone, Gianvito Vilé, Giovanni Di Liberto, Gianfranco Pacchioni
|
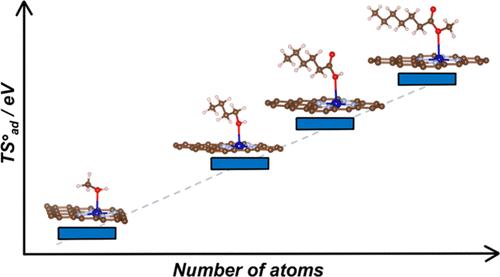
Single-atom catalysts (SACs) are rapidly gaining attention as a versatile class of materials that combine the advantages of both homogeneous and heterogeneous catalysis. A growing number of studies aim to identify potential new SACs or to describe their structure and reactivity through ab initio quantum chemical simulations. While many computational studies primarily address reactions involving small molecules, such as water splitting or CO2 reduction, the application scope of SACs is rapidly broadening to include the production of fine chemicals and the conversion of biomass-derived platform molecules, processes that involve larger, more complex reactants. Using density-functional theory (DFT) simulations, we demonstrate that, while an approximate treatment of entropy is acceptable for molecules with up to three atoms, it introduces substantial errors in reactions involving more complex molecules. Our results reveal a linear correlation between the entropy of adsorbed molecules and that of the corresponding isolated species, mirroring trends observed on extended catalytic surfaces. For the largest systems investigated in this study, the entropy of the free molecule is reduced by approximately 10–20% upon adsorption; for small molecules, this reduction can range from 50 to 70%. This disparity arises because, on SACs, the translational entropy is effectively zero, the rotational entropy is minimal, and the vibrational entropy increases with the size of the molecule. Moreover, the entropy of adsorbates scales linearly with the number of atoms in the molecule, allowing for the prediction of entropic contributions of adsorbates on SACs without additional computational cost. Using propyne hydrogenation as a test, we demonstrate that the reaction energy profile computed with current approximate approaches for estimating the entropy of adsorbates differs significantly from the profile where entropy is explicitly included. These findings highlight the importance of considering adsorbate entropy for accurately predicting the catalytic activity of SACs, particularly for reactions involving complex molecules.
中文翻译:

解码吸附物熵在单原子催化剂反应性中的作用
单原子催化剂 (SAC) 作为一类结合了均相和非均相催化优点的多功能材料,正迅速受到关注。越来越多的研究旨在通过 ab initio 量子化学模拟来识别潜在的新 SAC 或描述它们的结构和反应性。虽然许多计算研究主要涉及涉及小分子的反应,例如水分解或 CO2 还原,但 SAC 的应用范围正在迅速扩大,包括精细化学品的生产和生物质衍生平台分子的转化,这些过程涉及更大、更复杂的反应物。使用密度泛函理论 (DFT) 模拟,我们证明,虽然熵的近似处理对于最多具有三个原子的分子是可以接受的,但它在涉及更复杂分子的反应中引入了大量误差。我们的结果揭示了吸附分子的熵与相应的分离物质的熵之间的线性相关性,反映了在扩展催化表面上观察到的趋势。对于本研究中研究的最大系统,游离分子的熵在吸附时减少了约 10-20%;对于小分子,这种减少范围为 50% 到 70%。之所以出现这种差异,是因为在 SAC 上,平移熵实际上为零,旋转熵最小,并且振动熵随着分子大小的增加而增加。此外,吸附物的熵与分子中的原子数呈线性关系,因此无需额外的计算成本即可预测吸附物对 SAC 的熵贡献。 使用丙炔氢化作为测试,我们证明使用当前用于估计吸附物熵的近似方法计算的反应能量曲线与明确包含熵的曲线有很大不同。这些发现强调了考虑吸附物熵对于准确预测 SAC 催化活性的重要性,特别是对于涉及复杂分子的反应。
更新日期:2024-12-19
中文翻译:
解码吸附物熵在单原子催化剂反应性中的作用
单原子催化剂 (SAC) 作为一类结合了均相和非均相催化优点的多功能材料,正迅速受到关注。越来越多的研究旨在通过 ab initio 量子化学模拟来识别潜在的新 SAC 或描述它们的结构和反应性。虽然许多计算研究主要涉及涉及小分子的反应,例如水分解或 CO2 还原,但 SAC 的应用范围正在迅速扩大,包括精细化学品的生产和生物质衍生平台分子的转化,这些过程涉及更大、更复杂的反应物。使用密度泛函理论 (DFT) 模拟,我们证明,虽然熵的近似处理对于最多具有三个原子的分子是可以接受的,但它在涉及更复杂分子的反应中引入了大量误差。我们的结果揭示了吸附分子的熵与相应的分离物质的熵之间的线性相关性,反映了在扩展催化表面上观察到的趋势。对于本研究中研究的最大系统,游离分子的熵在吸附时减少了约 10-20%;对于小分子,这种减少范围为 50% 到 70%。之所以出现这种差异,是因为在 SAC 上,平移熵实际上为零,旋转熵最小,并且振动熵随着分子大小的增加而增加。此外,吸附物的熵与分子中的原子数呈线性关系,因此无需额外的计算成本即可预测吸附物对 SAC 的熵贡献。 使用丙炔氢化作为测试,我们证明使用当前用于估计吸附物熵的近似方法计算的反应能量曲线与明确包含熵的曲线有很大不同。这些发现强调了考虑吸附物熵对于准确预测 SAC 催化活性的重要性,特别是对于涉及复杂分子的反应。





























 京公网安备 11010802027423号
京公网安备 11010802027423号