npj Computational Materials ( IF 9.4 ) Pub Date : 2024-12-19 , DOI: 10.1038/s41524-024-01481-6 Simone Perego, Luigi Bonati
|
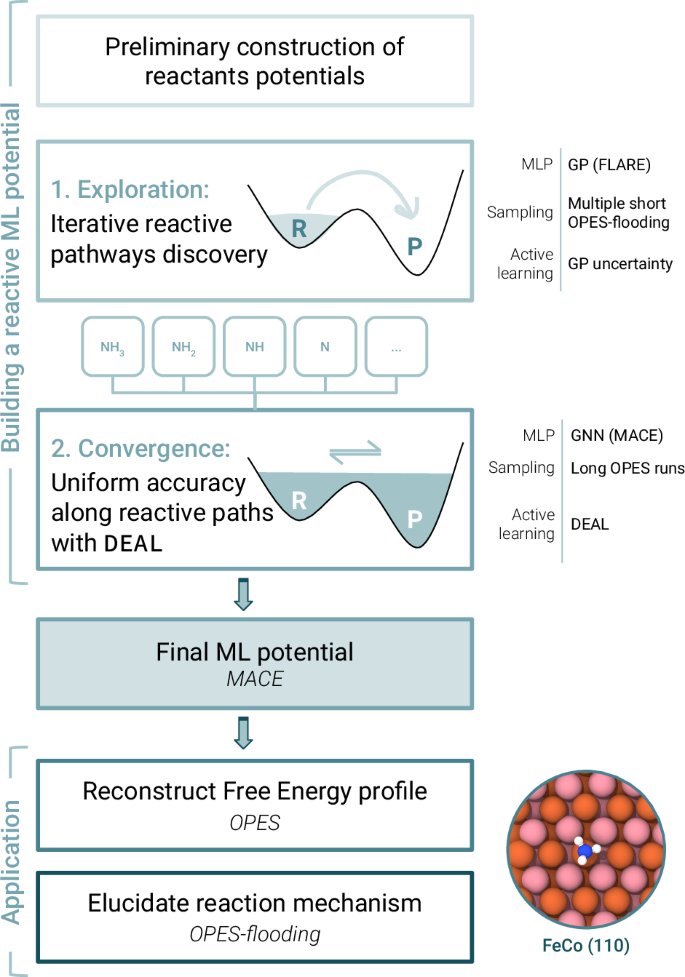
Simulating catalytic reactivity under operative conditions poses a significant challenge due to the dynamic nature of the catalysts and the high computational cost of electronic structure calculations. Machine learning potentials offer a promising avenue to simulate dynamics at a fraction of the cost, but they require datasets containing all relevant configurations, particularly reactive ones. Here, we present a scheme to construct reactive potentials in a data-efficient manner. This is achieved by combining enhanced sampling methods first with Gaussian processes to discover transition paths and then with graph neural networks to obtain a uniformly accurate description. The necessary configurations are extracted via a Data-Efficient Active Learning (DEAL) procedure based on local environment uncertainty. We validated our approach by studying several reactions related to the decomposition of ammonia on iron-cobalt alloy catalysts. Our scheme proved to be efficient, requiring only ~1000 DFT calculations per reaction, and robust, sampling reactive configurations from the different accessible pathways. Using this potential, we calculated free energy profiles and characterized reaction mechanisms, showing the ability to provide microscopic insights into complex processes under dynamic conditions.
中文翻译:
通过主动学习和增强采样对催化反应性进行建模的数据高效机器学习潜力
由于催化剂的动态性质和电子结构计算的高计算成本,在操作条件下模拟催化反应性是一项重大挑战。机器学习的潜力提供了一种很有前途的途径,可以以一小部分成本模拟动力学,但它们需要包含所有相关配置的数据集,尤其是反应式配置。在这里,我们提出了一种以数据高效的方式构建无功势的方案。这是通过首先将增强的采样方法与高斯过程相结合来发现过渡路径,然后与图形神经网络相结合以获得统一准确的描述来实现的。根据本地环境的不确定性,通过数据高效的主动学习 (DEAL) 程序提取必要的配置。我们通过研究与铁钴合金催化剂上氨分解相关的几个反应来验证我们的方法。我们的方案被证明是有效的,每个反应只需要 ~1000 次 DFT 计算,并且从不同的可访问途径中稳定地采样反应构型。利用这种电位,我们计算了自由能分布并表征了反应机理,展示了在动态条件下提供对复杂过程的微观见解的能力。





























 京公网安备 11010802027423号
京公网安备 11010802027423号