npj Computational Materials ( IF 9.4 ) Pub Date : 2024-12-19 , DOI: 10.1038/s41524-024-01444-x Hongwei Du, Jiamin Wang, Jian Hui, Lanting Zhang, Hong Wang
|
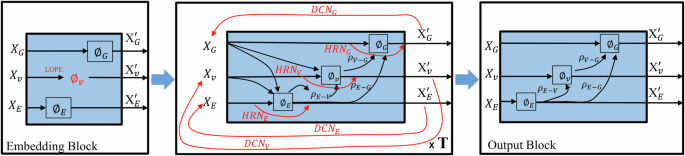
Modern generative models based on deep learning have made it possible to design millions of hypothetical materials. To screen these candidate materials and identify promising new materials, we need fast and accurate models to predict material properties. Graphical neural networks (GNNs) have become a current research focus due to their ability to directly act on the graphical representation of molecules and materials, enabling comprehensive capture of important information and showing excellent performance in predicting material properties. Nevertheless, GNNs still face several key problems in practical applications: First, although existing nested graph network strategies increase critical structural information such as bond angles, they significantly increase the number of trainable parameters in the model, resulting in a increase in training costs; Second, extending GNN models to broader domains such as molecules, crystalline materials, and catalysis, as well as adapting to small data sets, remains a challenge. Finally, the scalability of GNN models is limited by the over-smoothing problem. To address these issues, we propose the DenseGNN model, which combines Dense Connectivity Network (DCN), hierarchical node-edge-graph residual networks (HRN), and Local Structure Order Parameters Embedding (LOPE) strategies to create a universal, scalable, and efficient GNN model. We have achieved state-of-the-art performance (SOAT) on several datasets, including JARVIS-DFT, Materials Project, QM9, Lipop, FreeSolv, ESOL, and OC22, demonstrating the generality and scalability of our approach. By merging DCN and LOPE strategies into GNN models in computing, crystal materials, and molecules, we have improved the performance of models such as GIN, Schnet, and Hamnet on materials datasets such as Matbench. The LOPE strategy optimizes the embedding representation of atoms and allows our model to train efficiently with a minimal level of edge connections. This substantially reduces computational costs and shortens the time required to train large GNNs while maintaining accuracy. Our technique not only supports building deeper GNNs and avoids performance penalties experienced by other models, but is also applicable to a variety of applications that require large deep learning models. Furthermore, our study demonstrates that by using structural embeddings from pre-trained models, our model not only outperforms other GNNs in distinguishing crystal structures but also approaches the standard X-ray diffraction (XRD) method.
中文翻译:
DenseGNN:通用且可扩展的更深层图神经网络,用于晶体和分子的高性能性质预测
基于深度学习的现代生成模型使设计数百万种假设材料成为可能。为了筛选这些候选材料并确定有前途的新材料,我们需要快速准确的模型来预测材料特性。图形神经网络 (GNN) 已成为当前的研究重点,因为它们能够直接作用于分子和材料的图形表示,能够全面捕获重要信息,并在预测材料性能方面表现出优异的性能。尽管如此,GNN 在实际应用中仍面临几个关键问题:首先,虽然现有的嵌套图网络策略增加了键角等关键结构信息,但它们显着增加了模型中可训练参数的数量,导致训练成本增加;其次,将 GNN 模型扩展到更广泛的领域,如分子、晶体材料和催化,以及适应小型数据集,仍然是一个挑战。最后,GNN 模型的可扩展性受到过度平滑问题的限制。为了解决这些问题,我们提出了 DenseGNN 模型,该模型结合了密集连接网络 (DCN)、分层节点-边-图残差网络 (HRN) 和局部结构有序参数嵌入 (LOPE) 策略,以创建一个通用、可扩展且高效的 GNN 模型。我们已经在多个数据集上实现了最先进的性能 (SOAT),包括 JARVIS-DFT、Materials Project、QM9、Lipop、FreeSolv、ESOL 和 OC22,证明了我们方法的通用性和可扩展性。 通过将 DCN 和 LOPE 策略合并到计算、晶体材料和分子中的 GNN 模型中,我们提高了 GIN、Schnet 和 Hamnet 等模型在 Matbench 等材料数据集上的性能。LOPE 策略优化了原子的嵌入表示,并允许我们的模型以最少的边缘连接进行高效训练。这大大降低了计算成本,并缩短了训练大型 GNN 所需的时间,同时保持了准确性。我们的技术不仅支持构建更深的 GNN 并避免其他模型遇到的性能损失,而且还适用于需要大型深度学习模型的各种应用程序。此外,我们的研究表明,通过使用来自预训练模型的结构嵌入,我们的模型不仅在区分晶体结构方面优于其他 GNN,而且接近标准 X 射线衍射 (XRD) 方法。





























 京公网安备 11010802027423号
京公网安备 11010802027423号