Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Interactions and Responsive Locations of CO2-Responsive Surfactants in an Oil–Water–Surfactant System: Molecular Dynamics Simulation and Free Energy Perturbation
Langmuir ( IF 3.7 ) Pub Date : 2024-12-17 , DOI: 10.1021/acs.langmuir.4c03466 Xuantong Lei, Benjieming Liu, Zhangxin Chen
Langmuir ( IF 3.7 ) Pub Date : 2024-12-17 , DOI: 10.1021/acs.langmuir.4c03466 Xuantong Lei, Benjieming Liu, Zhangxin Chen
|
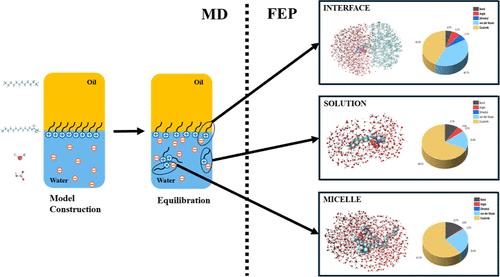
Understanding the mechanism of a CO2-responsive surfactant is essential for enhancing its industrial applications. Conventional experimental methods face challenges in pinpointing the exact location of proton transfer within the system and in accurately describing the impact of intermolecular and intramolecular interactions on the CO2 responsiveness of such substances. To address this gap, this study employs molecular dynamics simulations and free energy perturbation methods to investigate the proton transfer process between a CO2-responsive cationic surfactant N′-dodecyl-N,N-dimethylacetamidinium (DMAAH+) and its counterion bicarbonate ion at the oil–water interface and micelle surface and in the bulk aqueous phase. Molecular dynamics simulations identified potential locations for the proton transfer process within the system and elucidated the types of interactions contributing to changes in Gibbs free energy. Subsequently, free energy perturbation was employed to calculate Gibbs free energy changes associated with proton transfer at different locations. The respective contributions of various intramolecular and intermolecular interactions were then compared and analyzed. It has been revealed that the deprotonation process is not thermodynamically spontaneous at all three responsive locations. The proton transfer occurs more frequently at the oil–water interface than at the micelle surface and is less common in the bulk aqueous phase. The findings enhance our understanding of the fundamental mechanisms governing the responsiveness of CO2-responsive surfactants and provide valuable insights for their practical application in industrial processes.
中文翻译:

油-水-表面活性剂系统中 CO2 响应性表面活性剂的分子相互作用和响应位置:分子动力学模拟和自由能扰动
了解 CO2 响应性表面活性剂的机理对于增强其工业应用至关重要。传统的实验方法在确定系统内质子转移的确切位置以及准确描述分子间和分子内相互作用对此类物质的 CO2 响应性的影响方面面临挑战。为了解决这一差距,本研究采用分子动力学模拟和自由能扰动方法研究了 CO2 响应性阳离子表面活性剂 N′-十二烷基-N,N-二甲基乙酰脒 (DMAAH+) 与其对离子碳酸氢根离子在油-水界面和胶束表面以及本体水相中的质子转移过程。分子动力学模拟确定了系统内质子转移过程的潜在位置,并阐明了导致吉布斯自由能变化的相互作用类型。随后,采用自由能扰动来计算与不同位置的质子转移相关的吉布斯自由能变化。然后比较和分析了各种分子内和分子间相互作用的各自贡献。已经揭示,去质子化过程在所有三个响应位置都不是热力学自发的。质子转移发生在油-水界面处比在胶束表面更频繁,并且在本体水相中不太常见。这些发现增强了我们对控制 CO2 响应性表面活性剂响应性的基本机制的理解,并为它们在工业过程中的实际应用提供了有价值的见解。
更新日期:2024-12-18
中文翻译:
油-水-表面活性剂系统中 CO2 响应性表面活性剂的分子相互作用和响应位置:分子动力学模拟和自由能扰动
了解 CO2 响应性表面活性剂的机理对于增强其工业应用至关重要。传统的实验方法在确定系统内质子转移的确切位置以及准确描述分子间和分子内相互作用对此类物质的 CO2 响应性的影响方面面临挑战。为了解决这一差距,本研究采用分子动力学模拟和自由能扰动方法研究了 CO2 响应性阳离子表面活性剂 N′-十二烷基-N,N-二甲基乙酰脒 (DMAAH+) 与其对离子碳酸氢根离子在油-水界面和胶束表面以及本体水相中的质子转移过程。分子动力学模拟确定了系统内质子转移过程的潜在位置,并阐明了导致吉布斯自由能变化的相互作用类型。随后,采用自由能扰动来计算与不同位置的质子转移相关的吉布斯自由能变化。然后比较和分析了各种分子内和分子间相互作用的各自贡献。已经揭示,去质子化过程在所有三个响应位置都不是热力学自发的。质子转移发生在油-水界面处比在胶束表面更频繁,并且在本体水相中不太常见。这些发现增强了我们对控制 CO2 响应性表面活性剂响应性的基本机制的理解,并为它们在工业过程中的实际应用提供了有价值的见解。





























 京公网安备 11010802027423号
京公网安备 11010802027423号