当前位置:
X-MOL 学术
›
Adv. Energy Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Water Electrooxidation Kinetics Clarified by Time‐Resolved X‐Ray Absorption and Electrochemical Impedance Spectroscopy for a Bulk‐Active Cobalt Material
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2024-12-18 , DOI: 10.1002/aenm.202403818 Shima Farhoosh, Si Liu, Paul Beyer, Stefan Mebs, Michael Haumann, Holger Dau
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2024-12-18 , DOI: 10.1002/aenm.202403818 Shima Farhoosh, Si Liu, Paul Beyer, Stefan Mebs, Michael Haumann, Holger Dau
|
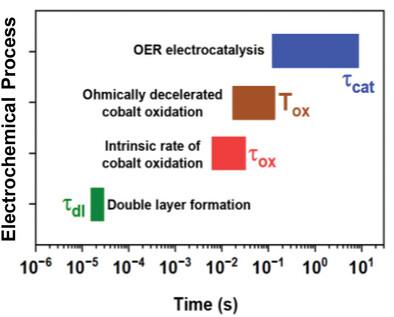
Water oxidation, the oxygen evolution reaction (OER), is the anodic process in electrocatalytic production of hydrogen and further green fuels. Transition‐metal oxyhydroxides with bulk‐phase OER activity of the complete material or amorphized near‐surface regions are of prime application interest, but their basic electrochemical properties are insufficiently understood. Here the timescale of functional processes is clarified by time‐resolved X‐ray absorption spectroscopy and electrochemical impedance spectroscopy (EIS) for a thickness‐series of cobalt oxyhydroxides films (about 35–550 nm). At the outer material surface, an electric double‐layer is formed in microseconds followed by clearly cobalt‐centered redox‐state changes of the bulk material in the low millisecond domain and a slow chemical step of O2 ‐formation, within hundreds of milliseconds. Conceptually interesting, the electrode potential likely controls the OER rate indirectly by driving the catalyst material to an increasingly oxidized state which promotes the rate‐limiting chemical step. Rate constants are derived for redox chemistry and catalysis from EIS data of low‐thickness catalyst films; at higher thicknesses, catalyst‐internal charge transport limitations become increasingly relevant. Relations between electrochemically active surface area, double‐layer capacitance, and redox (pseudo‐)capacitance are discussed. These results can increase the power of EIS analyses and support knowledge‐guided optimization of a broader class of OER catalyst materials.
中文翻译:

通过时间分辨 X 射线吸收和电化学阻抗谱阐明本体活性钴材料的水电氧化动力学
水氧化,即析氧反应 (OER),是电催化生产氢气和其他绿色燃料的阳极过程。具有完整材料或非晶化近表面区域的体相 OER 活性的过渡金属氢氧化物是主要应用兴趣,但对其基本电化学性质了解不足。在这里,通过时间分辨 X 射线吸收光谱和电化学阻抗谱 (EIS) 阐明了厚度系列钴羟基氧化物薄膜(约 35-550 nm)的功能过程的时间尺度。在材料外表面,在微秒内形成双电层,随后在低毫秒域中块状材料发生明显以钴为中心的氧化还原态变化,并在数百毫秒内形成 O2 的缓慢化学步骤。从概念上讲,电极电位可能通过将催化剂材料驱动到日益氧化的状态来间接控制 OER 速率,从而促进限速化学步骤。氧化还原化学和催化的速率常数是从低厚度催化剂膜的 EIS 数据中得出的;在更高的厚度下,催化剂内部电荷传输的限制变得越来越重要。讨论了电化学活性表面积、双电层电容和氧化还原(伪)电容之间的关系。这些结果可以提高 EIS 分析的能力,并支持对更广泛的 OER 催化剂材料进行知识指导的优化。
更新日期:2024-12-18
中文翻译:
通过时间分辨 X 射线吸收和电化学阻抗谱阐明本体活性钴材料的水电氧化动力学
水氧化,即析氧反应 (OER),是电催化生产氢气和其他绿色燃料的阳极过程。具有完整材料或非晶化近表面区域的体相 OER 活性的过渡金属氢氧化物是主要应用兴趣,但对其基本电化学性质了解不足。在这里,通过时间分辨 X 射线吸收光谱和电化学阻抗谱 (EIS) 阐明了厚度系列钴羟基氧化物薄膜(约 35-550 nm)的功能过程的时间尺度。在材料外表面,在微秒内形成双电层,随后在低毫秒域中块状材料发生明显以钴为中心的氧化还原态变化,并在数百毫秒内形成 O2 的缓慢化学步骤。从概念上讲,电极电位可能通过将催化剂材料驱动到日益氧化的状态来间接控制 OER 速率,从而促进限速化学步骤。氧化还原化学和催化的速率常数是从低厚度催化剂膜的 EIS 数据中得出的;在更高的厚度下,催化剂内部电荷传输的限制变得越来越重要。讨论了电化学活性表面积、双电层电容和氧化还原(伪)电容之间的关系。这些结果可以提高 EIS 分析的能力,并支持对更广泛的 OER 催化剂材料进行知识指导的优化。
































 京公网安备 11010802027423号
京公网安备 11010802027423号