当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular modeling of hydrogen adsorption and mobility in transition metal-functionalized polycrystalline CNTs using an energy-centered approach
Applied Surface Science ( IF 6.3 ) Pub Date : 2024-12-17 , DOI: 10.1016/j.apsusc.2024.162121 Saurabh Mishra, Fan Yang, S.I. Kundalwal
Applied Surface Science ( IF 6.3 ) Pub Date : 2024-12-17 , DOI: 10.1016/j.apsusc.2024.162121 Saurabh Mishra, Fan Yang, S.I. Kundalwal
|
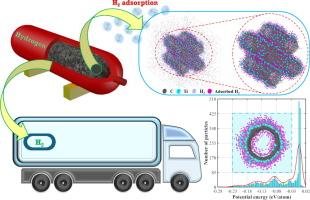
Despite notable progress in synthesizing large-sized carbon nanotubes (CNTs), persisting challenges arise from their inherent polycrystallinity and tendency to agglomerate. The investigation focuses on the interactions of molecular hydrogen with polycrystalline CNTs (PCNTs) functionalized with various concentrations of transition metals (Fe, Ni, and Ti) through molecular dynamics simulations (MDS). A novel potential energy distribution (PED) technique, integrated with grand canonical Monte Carlo (GCMC) simulations, was employed to identify and calculate the adsorbed hydrogen. Comparative analysis revealed that Ti-functionalized PCNTs (Ti-PCNTs) exhibited superior hydrogen adsorption capacity, characterized by deep local minima in PED with broad distribution, outperforming Fe and Ni-PCNTs. At 100 bar, 10 at% Ti-PCNT exhibits the highest hydrogen storage capacity, with a maximum of 7.14 wt% at 200 K and 5.2 wt% at 300 K. In comparison, 10 at% Ni-PCNT and Fe-PCNT attain maximum gravimetric densities of 3.31 wt% (1.75 wt%) and 3.19 wt% (1.7 wt%) at 200 K (300 K), respectively. Notably, bundled Ti-PCNTs (intertube spacing: 10 Å) achieved a substantial hydrogen gravimetric density of 6.6 wt% at 300 K and 100 bar, exceeding the US Department of Energy’s targets. Subsequently, the average adsorption energies were calculated as 0.115 eV/H2 and 0.110 eV/H2 for isolated and bundled Ti-PCNTs at 200 K, increasing to 0.128 eV/H2 and 0.123 eV/H2 at 300 K, indicating stable and stronger adsorption states relative to undoped PCNTs. Furthermore, functionalization reduced hydrogen mobility, as Ti-PCNTs exhibited lower diffusion coefficients compared to Ni and Fe-PCNTs. Specifically, the diffusion coefficients for a 10 at% Ti-PCNT bundle were calculated as 9.47 × 10-8 m2/s and 1.85 × 10-7 m2/s at 200 K and 300 K, respectively. Ultimately, the integrated PED-MDS approach demonstrated robustness in evaluating adsorption metrics, endorsing Ti-PCNT as a promising candidate for efficient hydrogen storage. The proposed computational framework will facilitate the development and optimization of novel materials that could further improve hydrogen storage performance.
中文翻译:

使用能量为中心的方法对过渡金属功能化多晶碳纳米管中的氢吸附和迁移率进行分子建模
尽管在合成大尺寸碳纳米管 (CNT) 方面取得了显著进展,但其固有的多晶性和团聚倾向带来了持续的挑战。该研究的重点是氢分子与通过分子动力学模拟 (MDS) 用各种浓度的过渡金属(Fe、Ni 和 Ti)功能化的多晶 CNT (PCNT) 的相互作用。采用一种新的势能分布 (PED) 技术,与大正则蒙特卡洛 (GCMC) 模拟相结合,用于识别和计算吸附的氢。比较分析表明,Ti 功能化 PCNT (Ti-PCNTs) 表现出优异的氢吸附能力,其特征是 PED 中具有较深的局部最小值,分布广泛,优于 Fe 和 Ni-PCNTs。在 100 bar 时,10 at% Ti-PCNT 表现出最高的储氢能力,在 200 K 时最大为 7.14 wt%,在 300 K 时最大为 5.2 wt%。相比之下,10 at% Ni-PCNT 和 Fe-PCNT 在 200 K (300 K) 时分别达到 3.31 wt% (1.75 wt%) 和 3.19 wt% (1.7 wt%) 的最大重量密度。值得注意的是,成束 Ti-PCNT(管间间距:10 Å)在 300 K 和 100 bar 下实现了 6.6 wt% 的氢气重量密度,超过了美国能源部的目标。随后,计算出分离和捆绑的 Ti-PCNT 在 200 K 时的平均吸附能为 0.115 eV/H2 和 0.110 eV/H2,在 300 K 时增加到 0.128 eV/H2 和 0.123 eV/H2,表明相对于未掺杂的 PCNT 具有稳定和更强的吸附状态。此外,功能化降低了氢迁移率,因为与 Ni 和 Fe-PCNT 相比,Ti-PCNT 表现出较低的扩散系数。 具体来说,在 200 K 和 300 K 时,10 at% Ti-PCNT 束的扩散系数分别为 9.47 × 10-8 m2/s 和 1.85 × 10-7 m2/s。最终,集成的 PED-MDS 方法在评估吸附指标方面表现出稳健性,认可 Ti-PCNT 是高效储氢的有前途的候选者。拟议的计算框架将促进新型材料的开发和优化,从而进一步提高储氢性能。
更新日期:2024-12-18
中文翻译:
使用能量为中心的方法对过渡金属功能化多晶碳纳米管中的氢吸附和迁移率进行分子建模
尽管在合成大尺寸碳纳米管 (CNT) 方面取得了显著进展,但其固有的多晶性和团聚倾向带来了持续的挑战。该研究的重点是氢分子与通过分子动力学模拟 (MDS) 用各种浓度的过渡金属(Fe、Ni 和 Ti)功能化的多晶 CNT (PCNT) 的相互作用。采用一种新的势能分布 (PED) 技术,与大正则蒙特卡洛 (GCMC) 模拟相结合,用于识别和计算吸附的氢。比较分析表明,Ti 功能化 PCNT (Ti-PCNTs) 表现出优异的氢吸附能力,其特征是 PED 中具有较深的局部最小值,分布广泛,优于 Fe 和 Ni-PCNTs。在 100 bar 时,10 at% Ti-PCNT 表现出最高的储氢能力,在 200 K 时最大为 7.14 wt%,在 300 K 时最大为 5.2 wt%。相比之下,10 at% Ni-PCNT 和 Fe-PCNT 在 200 K (300 K) 时分别达到 3.31 wt% (1.75 wt%) 和 3.19 wt% (1.7 wt%) 的最大重量密度。值得注意的是,成束 Ti-PCNT(管间间距:10 Å)在 300 K 和 100 bar 下实现了 6.6 wt% 的氢气重量密度,超过了美国能源部的目标。随后,计算出分离和捆绑的 Ti-PCNT 在 200 K 时的平均吸附能为 0.115 eV/H2 和 0.110 eV/H2,在 300 K 时增加到 0.128 eV/H2 和 0.123 eV/H2,表明相对于未掺杂的 PCNT 具有稳定和更强的吸附状态。此外,功能化降低了氢迁移率,因为与 Ni 和 Fe-PCNT 相比,Ti-PCNT 表现出较低的扩散系数。 具体来说,在 200 K 和 300 K 时,10 at% Ti-PCNT 束的扩散系数分别为 9.47 × 10-8 m2/s 和 1.85 × 10-7 m2/s。最终,集成的 PED-MDS 方法在评估吸附指标方面表现出稳健性,认可 Ti-PCNT 是高效储氢的有前途的候选者。拟议的计算框架将促进新型材料的开发和优化,从而进一步提高储氢性能。





























 京公网安备 11010802027423号
京公网安备 11010802027423号