当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Dissociative mechanism from NH[formula omitted] and CH[formula omitted] on Ni-doped graphene: Tuning electronic and optical properties
Applied Surface Science ( IF 6.3 ) Pub Date : 2024-12-14 , DOI: 10.1016/j.apsusc.2024.162022 Amil Aligayev, U. Jabbarli, U. Samadova, F.J. Dominguez–Gutierrez, S. Papanikolaou, Qing Huang
Applied Surface Science ( IF 6.3 ) Pub Date : 2024-12-14 , DOI: 10.1016/j.apsusc.2024.162022 Amil Aligayev, U. Jabbarli, U. Samadova, F.J. Dominguez–Gutierrez, S. Papanikolaou, Qing Huang
|
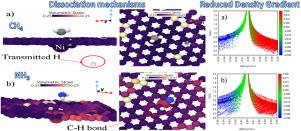
In this study, we employ a multi-scale computational modeling approach, combining density functional theory (DFT) and self-consistent charge density functional tight binding (SCC-DFTB), to investigate hydrogen (H2 3 4 4 3 4 3 3 2 2 4
中文翻译:

NH[公式省略] 和 CH[公式省略] 对镍掺杂石墨烯的解离机制:调整电子和光学特性
在这项研究中,我们采用多尺度计算建模方法,结合密度泛函理论 (DFT) 和自洽电荷密度泛函紧密结合 (SCC-DFTB),研究氨 (NH3) 和甲烷 (CH4) 在原始和镍掺杂石墨烯上的氢 (H2) 生产和解离机制。这些二维材料在高级气体传感和催化方面具有巨大的应用潜力。我们的分析表明,通过功函数计算验证的 Ni 掺杂石墨烯是一种很有前途的气体分离和制氢材料。通过 DFT 计算计算化学势、化学硬度、电负性、亲电性、振动频率、吸附和吉布斯能量,对吸附分子的样品进行表征。甲烷分子优先吸附在石墨烯的六方环中心,而氨与碳原子的相互作用更强烈,突出了 CH4 和 NH3 的不同分子掺杂机制。动力学模拟表明,CH4 分裂成 CH3+H,Ni 掺杂石墨烯有助于增强氢传输,而 NH3 解离成 NH2+H,这可能导致 N2H4 的形成。我们的非平衡格林函数 (NEGF) 模拟表明,在气体相互作用期间,氢原子在掺杂镍石墨烯上的传输增加。这些发现表明,Ni 掺杂石墨烯在气体分离、制氢和高灵敏度传感器方面的应用优于原始石墨烯。
更新日期:2024-12-14
中文翻译:
NH[公式省略] 和 CH[公式省略] 对镍掺杂石墨烯的解离机制:调整电子和光学特性
在这项研究中,我们采用多尺度计算建模方法,结合密度泛函理论 (DFT) 和自洽电荷密度泛函紧密结合 (SCC-DFTB),研究氨 (NH3) 和甲烷 (CH4) 在原始和镍掺杂石墨烯上的氢 (H2) 生产和解离机制。这些二维材料在高级气体传感和催化方面具有巨大的应用潜力。我们的分析表明,通过功函数计算验证的 Ni 掺杂石墨烯是一种很有前途的气体分离和制氢材料。通过 DFT 计算计算化学势、化学硬度、电负性、亲电性、振动频率、吸附和吉布斯能量,对吸附分子的样品进行表征。甲烷分子优先吸附在石墨烯的六方环中心,而氨与碳原子的相互作用更强烈,突出了 CH4 和 NH3 的不同分子掺杂机制。动力学模拟表明,CH4 分裂成 CH3+H,Ni 掺杂石墨烯有助于增强氢传输,而 NH3 解离成 NH2+H,这可能导致 N2H4 的形成。我们的非平衡格林函数 (NEGF) 模拟表明,在气体相互作用期间,氢原子在掺杂镍石墨烯上的传输增加。这些发现表明,Ni 掺杂石墨烯在气体分离、制氢和高灵敏度传感器方面的应用优于原始石墨烯。
































 京公网安备 11010802027423号
京公网安备 11010802027423号