当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
CGPDTA: An Explainable Transfer Learning‐Based Predictor With Molecule Substructure Graph for Drug‐Target Binding Affinity
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-12-10 , DOI: 10.1002/jcc.27538 Qing Fan, Yingxu Liu, Simeng Zhang, Xiangzhen Ning, Chengcheng Xu, Weijie Han, Yanmin Zhang, Yadong Chen, Jun Shen, Haichun Liu
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-12-10 , DOI: 10.1002/jcc.27538 Qing Fan, Yingxu Liu, Simeng Zhang, Xiangzhen Ning, Chengcheng Xu, Weijie Han, Yanmin Zhang, Yadong Chen, Jun Shen, Haichun Liu
|
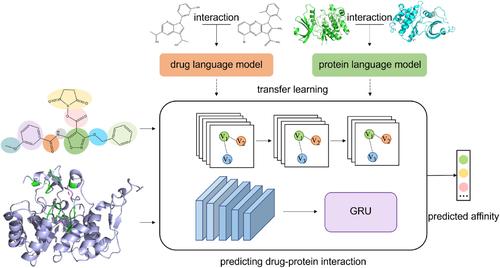
Identifying interactions between drugs and targets is crucial for drug discovery and development. Nevertheless, the determination of drug‐target binding affinities (DTAs) through traditional experimental methods is a time‐consuming process. Conventional approaches to predicting drug‐target interactions (DTIs) frequently prove inadequate due to an insufficient representation of drugs and targets, resulting in ineffective feature capture and questionable interpretability of results. To address these challenges, we introduce CGPDTA, a novel deep learning framework empowered by transfer learning, designed explicitly for the accurate prediction of DTAs. CGPDTA leverages the complementarity of drug–drug and protein–protein interaction knowledge through advanced drug and protein language models. It further enhances predictive capability and interpretability by incorporating molecular substructure graphs and protein pocket sequences to represent local features of drugs and targets effectively. Our findings demonstrate that CGPDTA not only outperforms existing methods in accuracy but also provides meaningful insights into the predictive process, marking a significant advancement in the field of drug discovery.
中文翻译:

CGPDTA:一种基于迁移学习的可解释预测器,具有药物靶标结合亲和力的分子子结构图
确定药物和靶标之间的相互作用对于药物发现和开发至关重要。然而,通过传统实验方法测定药物靶标结合亲和力 (DTA) 是一个耗时的过程。由于药物和靶点的表示不足,预测药物-靶点相互作用 (DTI) 的传统方法经常被证明是不够的,导致特征捕获无效和结果的可解释性有问题。为了应对这些挑战,我们引入了 CGPDTA,这是一种由迁移学习提供支持的新型深度学习框架,专为准确预测 DTA 而设计。CGPDTA 通过先进的药物和蛋白质语言模型利用药物-药物和蛋白质-蛋白质相互作用知识的互补性。它通过结合分子亚结构图和蛋白质口袋序列来有效地表示药物和靶点的局部特征,从而进一步增强预测能力和可解释性。我们的研究结果表明,CGPDTA 不仅在准确性上优于现有方法,而且还为预测过程提供了有意义的见解,标志着药物发现领域的重大进步。
更新日期:2024-12-10
中文翻译:
CGPDTA:一种基于迁移学习的可解释预测器,具有药物靶标结合亲和力的分子子结构图
确定药物和靶标之间的相互作用对于药物发现和开发至关重要。然而,通过传统实验方法测定药物靶标结合亲和力 (DTA) 是一个耗时的过程。由于药物和靶点的表示不足,预测药物-靶点相互作用 (DTI) 的传统方法经常被证明是不够的,导致特征捕获无效和结果的可解释性有问题。为了应对这些挑战,我们引入了 CGPDTA,这是一种由迁移学习提供支持的新型深度学习框架,专为准确预测 DTA 而设计。CGPDTA 通过先进的药物和蛋白质语言模型利用药物-药物和蛋白质-蛋白质相互作用知识的互补性。它通过结合分子亚结构图和蛋白质口袋序列来有效地表示药物和靶点的局部特征,从而进一步增强预测能力和可解释性。我们的研究结果表明,CGPDTA 不仅在准确性上优于现有方法,而且还为预测过程提供了有意义的见解,标志着药物发现领域的重大进步。
































 京公网安备 11010802027423号
京公网安备 11010802027423号