当前位置:
X-MOL 学术
›
Dalton Trans.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
High-pressure phase transition and amorphization of BaV2O6
Dalton Transactions ( IF 3.5 ) Pub Date : 2024-12-09 , DOI: 10.1039/d4dt03091e
Peijie Zhang, Pablo Botella, Neha Bura, Jose-Luis Rodrigo, Josu Sanchez-Martin, David Vie, Catalin Popescu, Daniel Errandonea
Dalton Transactions ( IF 3.5 ) Pub Date : 2024-12-09 , DOI: 10.1039/d4dt03091e
Peijie Zhang, Pablo Botella, Neha Bura, Jose-Luis Rodrigo, Josu Sanchez-Martin, David Vie, Catalin Popescu, Daniel Errandonea
|
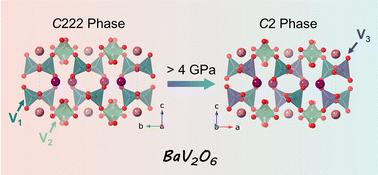
The structural evolution of metavanadate compounds under high pressure offers valuable insights into phase transitions and changes in material properties. This study explores the structural behavior of BaV2O6 under pressures up to 12 GPa using powder X-ray diffraction and density-functional theory (DFT) simulations. The results indicate a phase transition from the ambient pressure orthorhombic phase (space group C222) to a monoclinic phase (space group C2) at 4 GPa, likely driven by the distortion of the vanadium oxide polyhedron. Above 10 GPa, the C2 phase undergoes amorphization, attributed to the breakdown of the infinite [VO4] chains into [VO3]− units. Additionally, BaV2O6 exhibits anisotropic lattice contraction and a relatively low bulk modulus (B0 ≈ 50 GPa). DFT calculations further explore the pressure dependence of enthalpy differences, Raman modes, and band structures, providing insights into the structural and electronic transformations of BaV2O6 under high pressure. This work deepens the understanding of the structural and band structure development of the metavanadate family under high pressure, contributing to advancements in materials science under extreme conditions.
中文翻译:

BaV2O6 的高压相变和非晶化
偏钒酸盐化合物在高压下的结构演变为了解相变和材料性能变化提供了有价值的见解。本研究使用粉末 X 射线衍射和密度泛函理论 (DFT) 模拟探讨了 BaV2O6 在高达 12 GPa 的压力下的结构行为。结果表明,在 4 GPa 处从环境压力正交相(空间群 C222)到单斜相(空间群 C2)的相变,这可能是由氧化钒多面体的变形驱动的。高于 10 GPa 时,C2 相发生非晶化,这归因于无限 [VO4] 链分解成 [VO3]− 单位。此外,BaV2O6 表现出各向异性晶格收缩和相对较低的体积模量 (B0 ≈ 50 GPa)。DFT 计算进一步探索了焓差、拉曼模式和能带结构的压力依赖性,为深入了解 BaV2O6 在高压下的结构和电子转变提供了见解。这项工作加深了对高压下偏钒酸盐家族结构和能带结构发展的理解,有助于极端条件下材料科学的进步。
更新日期:2024-12-09
中文翻译:
BaV2O6 的高压相变和非晶化
偏钒酸盐化合物在高压下的结构演变为了解相变和材料性能变化提供了有价值的见解。本研究使用粉末 X 射线衍射和密度泛函理论 (DFT) 模拟探讨了 BaV2O6 在高达 12 GPa 的压力下的结构行为。结果表明,在 4 GPa 处从环境压力正交相(空间群 C222)到单斜相(空间群 C2)的相变,这可能是由氧化钒多面体的变形驱动的。高于 10 GPa 时,C2 相发生非晶化,这归因于无限 [VO4] 链分解成 [VO3]− 单位。此外,BaV2O6 表现出各向异性晶格收缩和相对较低的体积模量 (B0 ≈ 50 GPa)。DFT 计算进一步探索了焓差、拉曼模式和能带结构的压力依赖性,为深入了解 BaV2O6 在高压下的结构和电子转变提供了见解。这项工作加深了对高压下偏钒酸盐家族结构和能带结构发展的理解,有助于极端条件下材料科学的进步。






























 京公网安备 11010802027423号
京公网安备 11010802027423号