当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Unveiling the molecular mechanism of Mn and Zn-catalyzed Ullmann-type C–O cross-coupling reactions
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-12-05 , DOI: 10.1039/d4cp02777a C. Rajalakshmi, Parvathi Santhoshkumar, Lydia Elizabeth Mathews, Ann Miriam Abraham, K. R. Rohit, Gopinathan Anilkumar, Vibin Ipe Thomas
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2024-12-05 , DOI: 10.1039/d4cp02777a C. Rajalakshmi, Parvathi Santhoshkumar, Lydia Elizabeth Mathews, Ann Miriam Abraham, K. R. Rohit, Gopinathan Anilkumar, Vibin Ipe Thomas
|
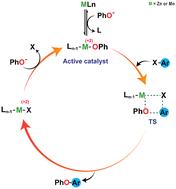
A detailed theoretical study delving into the molecular mechanisms of the Ullmann-type O-arylation reactions catalyzed by manganese and zinc metal ions has been investigated with the aid of the density functional theory (DFT) method. In contrast to the redox-active mechanisms proposed for classical Ullmann-type condensation reaction, a redox-neutral mechanism involving σ-bond metathesis emerged as the most appealing pathway for the investigated high-valent Mn(II) and Zn(II)-catalyzed O-arylation reactions. The mechanism remains invariant with respect to the nature of the central metal, ligand, base, etc. This unusuality in the mechanism has been dissected by considering three cases: ligand-free and ligand-assisted Mn(II)-catalyzed O-arylation reaction and ligand-assisted Zn(II)-catalyzed O-arylation reactions. In each class, a metal phenoxide species was identified as the active catalyst. The unusual mechanistic trends observed in these C–O cross-coupling reactions could be attributed to the stable electronic configurations (d5, d10) combined with the higher oxidation states of the catalytic metal centers (Mn(II), Zn(II)). The exploration into the electronic effects of functional groups in controlling the reaction feasibility within each metal variant disclosed a consistent trend irrespective of the transition metal catalyst involved in the reaction. It was found that the introduction of electron-withdrawing groups at the para-position of organic halide lowers the energy of their lowest unoccupied molecular orbitals (LUMO), thereby lowering the HOMO–LUMO gap between the coupling partners. This study thereby revealed a comprehensive understanding of the fundamental mechanisms exhibited by high-valent first-row transition metal catalysts that could foster the development of eco-friendly protocols for preparing biaryl ether moieties.
中文翻译:

揭示 Mn 和 Zn 催化的 Ullmann 型 C-O 交叉偶联反应的分子机制
借助密度泛函理论 (DFT) 方法,深入研究了由锰和锌金属离子催化的 Ullmann 型 O-芳基化反应的分子机制的详细理论研究。与经典的 Ullmann 型缩合反应提出的氧化还原活性机制相反,涉及 σ 键复分解的氧化还原中性机制成为所研究的高价 Mn(II) 和 Zn(II) 催化的 O-芳基化反应最吸引人的途径。该机制就中心金属、配体、碱等的性质保持不变。通过考虑三种情况,已经剖析了机制中的这种不寻常性:无配体和配体辅助 Mn(II) 催化的 O-芳基化反应和配体辅助的 Zn(II) 催化的 O-芳基化反应。在每类中,一种金属苯氧化物被鉴定为活性催化剂。在这些 C-O 交叉偶联反应中观察到的不寻常的机理趋势可归因于稳定的电子构型 (d5, d10) 以及催化金属中心(Mn(II)、Zn(II)) 的较高氧化态。对官能团在控制每种金属变体中的反应可行性方面的电子效应的探索揭示了与反应中涉及的过渡金属催化剂无关的一致趋势。研究发现,在有机卤化物的对位引入吸电子基团降低了它们最低未占据分子轨道 (LUMO) 的能量,从而降低了耦合伙伴之间的 HOMO-LUMO 间隙。 因此,本研究揭示了对高价第一行过渡金属催化剂所表现出的基本机制的全面理解,这些机制可以促进制备联芳基醚部分的环保方案的开发。
更新日期:2024-12-05
中文翻译:
揭示 Mn 和 Zn 催化的 Ullmann 型 C-O 交叉偶联反应的分子机制
借助密度泛函理论 (DFT) 方法,深入研究了由锰和锌金属离子催化的 Ullmann 型 O-芳基化反应的分子机制的详细理论研究。与经典的 Ullmann 型缩合反应提出的氧化还原活性机制相反,涉及 σ 键复分解的氧化还原中性机制成为所研究的高价 Mn(II) 和 Zn(II) 催化的 O-芳基化反应最吸引人的途径。该机制就中心金属、配体、碱等的性质保持不变。通过考虑三种情况,已经剖析了机制中的这种不寻常性:无配体和配体辅助 Mn(II) 催化的 O-芳基化反应和配体辅助的 Zn(II) 催化的 O-芳基化反应。在每类中,一种金属苯氧化物被鉴定为活性催化剂。在这些 C-O 交叉偶联反应中观察到的不寻常的机理趋势可归因于稳定的电子构型 (d5, d10) 以及催化金属中心(Mn(II)、Zn(II)) 的较高氧化态。对官能团在控制每种金属变体中的反应可行性方面的电子效应的探索揭示了与反应中涉及的过渡金属催化剂无关的一致趋势。研究发现,在有机卤化物的对位引入吸电子基团降低了它们最低未占据分子轨道 (LUMO) 的能量,从而降低了耦合伙伴之间的 HOMO-LUMO 间隙。 因此,本研究揭示了对高价第一行过渡金属催化剂所表现出的基本机制的全面理解,这些机制可以促进制备联芳基醚部分的环保方案的开发。





























 京公网安备 11010802027423号
京公网安备 11010802027423号