当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Chemical Bond Overlap Descriptors From Multiconfiguration Wavefunctions
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-11-28 , DOI: 10.1002/jcc.27534 Carlos V. Santos‐Jr, Elfi Kraka, Renaldo T. Moura
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2024-11-28 , DOI: 10.1002/jcc.27534 Carlos V. Santos‐Jr, Elfi Kraka, Renaldo T. Moura
|
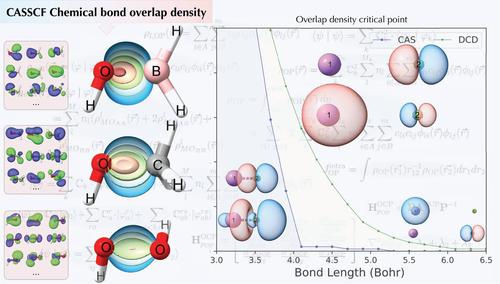
The chemical bond is a fundamental concept in chemistry, and various models and descriptors have evolved since the advent of quantum mechanics. This study extends the overlap density and its topological descriptors (OP/TOP) to multiconfigurational wavefunctions. We discuss a comparative analysis of OP/TOP descriptors using CASSCF and DCD‐CAS(2) wavefunctions for a diverse range of molecular systems, including X–O bonds in X–OH (XH, Li, Na, H2 B, H3 C, H2 N, HO, F) and Li–X′ (XF, Cl, and Br). Results show that OP/TOP aligns with bonding models like the quantum theory of atoms in molecules (QTAIM) and local vibrational modes theory, revealing insights such as overlap densities shifting towards the more electronegative atom in polar bonds. The Li–F dissociation profile using OP/TOP descriptors demonstrated sensitivity to ionic/neutral inversion during Li–F dissociation, highlighting their potential for elucidating complex bond phenomena and offering new avenues for understanding multiconfigurational chemical bond dynamics.
中文翻译:

来自多配置 Wavefunctions 的化学键重叠描述符
化学键是化学中的一个基本概念,自量子力学出现以来,各种模型和描述符已经发展起来。本研究将重叠密度及其拓扑描述符 (OP/TOP) 扩展到多构型波函数。我们讨论了使用 CASSCF 和 DCD-CAS(2) 波函数对各种分子系统进行 OP/TOP 描述符的比较分析,包括 X-OH(XH、Li、Na、H2B、H3C、H2N、HO、F)和 Li-X′(XF、Cl 和 Br)中的 X-O 键。结果表明,OP/TOP 与分子中原子的量子理论 (QTAIM) 和局部振动模式理论等键合模型一致,揭示了诸如重叠密度向极性键中电负性更高的原子转移等见解。使用 OP/TOP 描述符的 Li-F 解离谱证明了对 Li-F 解离过程中离子/中性反转的敏感性,突出了它们阐明复杂键现象的潜力,并为理解多构化学键动力学提供了新的途径。
更新日期:2024-11-28
中文翻译:
来自多配置 Wavefunctions 的化学键重叠描述符
化学键是化学中的一个基本概念,自量子力学出现以来,各种模型和描述符已经发展起来。本研究将重叠密度及其拓扑描述符 (OP/TOP) 扩展到多构型波函数。我们讨论了使用 CASSCF 和 DCD-CAS(2) 波函数对各种分子系统进行 OP/TOP 描述符的比较分析,包括 X-OH(XH、Li、Na、H2B、H3C、H2N、HO、F)和 Li-X′(XF、Cl 和 Br)中的 X-O 键。结果表明,OP/TOP 与分子中原子的量子理论 (QTAIM) 和局部振动模式理论等键合模型一致,揭示了诸如重叠密度向极性键中电负性更高的原子转移等见解。使用 OP/TOP 描述符的 Li-F 解离谱证明了对 Li-F 解离过程中离子/中性反转的敏感性,突出了它们阐明复杂键现象的潜力,并为理解多构化学键动力学提供了新的途径。






























 京公网安备 11010802027423号
京公网安备 11010802027423号