当前位置:
X-MOL 学术
›
Adv. Synth. Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
From gem-Dichlorocyclobutenones to Cyclobutenols: Unveiling a Ruthenium-Catalyzed Allylic Reduction-Asymmetric Transfer Hydrogenation Cascade
Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2024-11-27 , DOI: 10.1002/adsc.202401406 Olivier Charron, Marharyta Kosiuha, Virginie Ratovelomanana-Vidal, Phannarath Phansavath, Geoffrey Gontard, Christophe Meyer
Advanced Synthesis & Catalysis ( IF 4.4 ) Pub Date : 2024-11-27 , DOI: 10.1002/adsc.202401406 Olivier Charron, Marharyta Kosiuha, Virginie Ratovelomanana-Vidal, Phannarath Phansavath, Geoffrey Gontard, Christophe Meyer
|
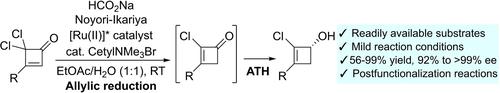
Cyclobutenones constitute an appealing class of substrates in catalytic asymmetric transformations leading to diversely substituted enantioenriched four-membered carbocycles, which are eliciting a growing interest in medicinal chemistry. Whilst several synthetically useful enantioselective conjugate addition reactions have been reported, the catalytic enantioselective reduction of the carbonyl group of simple cyclobutenones remains an elusive transformation. Herein, we disclose the discovery of a novel allylic reduction-asymmetric transfer hydrogenation cascade, catalyzed by a Noyori-Ikariya ruthenium complex, from readily available gem-dichlorocyclobutenones, leading to 2-chlorocyclobutenols with high optical purities, which can be engaged in postfunctionalization reactions enabling access to substituted four-membered rings.
中文翻译:

从 gem-二氯环丁烯酮到环丁烯醇:揭开钌催化烯丙基还原-不对称转移氢化级联的面纱
环丁烯酮在催化不对称转化中构成了一类有吸引力的底物,导致多种取代的对映体富集的四元碳环,这引起了人们对药物化学的日益增长的兴趣。虽然已经报道了几种合成有用的对映选择性偶联物加成反应,但简单环丁烯酮羰基的催化对映选择性还原仍然是一个难以捉摸的转化。在此,我们揭示了一种新的烯丙基还原-不对称转移氢化级联反应,由 Noyori-Ikariya 钌配合物催化,由现成的 gem-二氯环丁烯酮催化,从现成的 gem-二氯环丁烯酮中产生具有高光学纯度的 2-氯环丁烯醇,它可以参与功能后化反应,从而能够获得取代的四元环。
更新日期:2024-11-27
中文翻译:
从 gem-二氯环丁烯酮到环丁烯醇:揭开钌催化烯丙基还原-不对称转移氢化级联的面纱
环丁烯酮在催化不对称转化中构成了一类有吸引力的底物,导致多种取代的对映体富集的四元碳环,这引起了人们对药物化学的日益增长的兴趣。虽然已经报道了几种合成有用的对映选择性偶联物加成反应,但简单环丁烯酮羰基的催化对映选择性还原仍然是一个难以捉摸的转化。在此,我们揭示了一种新的烯丙基还原-不对称转移氢化级联反应,由 Noyori-Ikariya 钌配合物催化,由现成的 gem-二氯环丁烯酮催化,从现成的 gem-二氯环丁烯酮中产生具有高光学纯度的 2-氯环丁烯醇,它可以参与功能后化反应,从而能够获得取代的四元环。
































 京公网安备 11010802027423号
京公网安备 11010802027423号